Advancing Statistical Literacy in Eye Care: A Series for Enhanced Clinical Decision-Making
Part 3: Design and Analysis of Clinical Research in Eye and Vision StudiesPurpose. To provide comprehensive guidance on designing and analysing clinical research studies in ophthalmology and optometry, encompassing observational studies, non-randomised controlled trials, randomised controlled trials, and advanced trial methodologies including platform trials, adaptive designs, and decentralised trials. This article addresses the unique methodological challenges in eye and vision research while integrating contemporary frameworks such as the estimands approach for defining treatment effects and the ethical imperative of systematic reviews before initiating new trials.
Material and Methods. A comprehensive literature review was conducted using PubMed, Scopus, Web of Science, and Cochrane databases, focusing on study designs applicable to eye care research with emphasis on methodological innovations from 2019 to 2025. Simulated datasets representing common ophthalmic research scenarios were generated using Python to illustrate design principles and analytical approaches. International guidelines including ICH E9(R1), CONSORT 2025, and regulatory guidance from FDA and EMA were synthesised to provide current best-practice recommendations.
Results. Different study designs serve distinct purposes in advancing eye care knowledge. Observational studies excel at identifying associations and generating hypotheses but require careful consideration of confounding factors. Non-randomised controlled trials, strengthened by propensity score methods and target trial emulation frameworks, offer pragmatic evaluation when randomisation is not feasible. Randomised controlled trials remain the gold standard for establishing causality but benefit from modern innovations including adaptive designs, platform trials enabling evaluation of multiple treatments, and the estimands framework clarifying precisely what treatment effects trials measure. Sequential Multiple Assignment Randomised Trials address the clinical reality that optimal treatment often requires adaptive sequencing. Decentralised trial elements expand access while maintaining scientific rigour. Key considerations for eye research include accounting for inter-eye correlation, managing paired organ data, and addressing masking challenges in surgical interventions.
Conclusion. Successful clinical research in eye and vision research requires matching study design to research questions, available resources, and ethical considerations. Modern trial methodology offers sophisticated tools for generating robust evidence efficiently, from the estimands framework ensuring trials measure clinically meaningful effects to platform designs enabling adaptive evaluation of multiple interventions. By understanding the strengths and limitations of each design type and applying appropriate analytical methods, researchers can advance evidence-based eye care while reducing research waste.
Introduction
Clinical research in ophthalmology and optometry has undergone a methodological revolution over the past decade, moving far beyond the traditional two-arm parallel randomised controlled trial. This transformation encompasses sophisticated study designs addressing complex questions about disease mechanisms, diagnostic accuracy, and treatment efficacy, while the unique anatomical and physiological characteristics of the visual system present both opportunities and challenges for researchers. This third instalment in our statistical literacy series builds upon the foundational concepts of descriptive statistics (Part 1)1 and inferential testing (Part 2)2 to explore the practical aspects of designing and analysing clinical studies in eye care, with particular emphasis on contemporary methodological innovations that can enhance research efficiency and validity.
The landscape of ophthalmic research encompasses diverse study designs, each suited to specific research questions and contexts. Observational studies have illuminated the natural history of diseases like age-related macular degeneration and identified risk factors for conditions such as myopic progression.3,4,5 Non-randomised controlled trials have evaluated interventions in real-world settings where randomisation poses ethical or practical challenges. Randomised controlled trials have established the efficacy of treatments ranging from anti-VEGF therapy for retinal diseases to surgical techniques for glaucoma. However, the field now benefits from methodological advances that extend well beyond these traditional approaches, including platform trials that evaluate multiple treatments within shared infrastructure, adaptive designs that modify trial parameters based on accumulating evidence, and the estimands framework that ensures trials measure clinically meaningful treatment effects.
Despite advances in ophthalmic research, persistent methodological challenges remain. Reviews of eye and vision randomised controlled trials have documented frequent shortcomings in trial design and statistical analysis. These shortcomings include incomplete or inappropriate sample size and power calculations, and insufficient adjustment for correlated eye data in two-eye study designs. Such deficiencies undermine the validity of inferences.6 These deficiencies not only waste resources but may lead to incorrect conclusions that affect patient care. The problem extends beyond individual studies: approximately 85 % of research investment globally is estimated to be wasted due to asking irrelevant questions, flawed design, non-publication, and poor reporting.7 In eye and vision research specifically, only 22.4 % of phase III trials cite systematic reviews as justification for the study, missing opportunities to build on existing evidence.8 Furthermore, the paired nature of eyes, the subjective nature of many visual outcomes, and the difficulty of masking surgical interventions create unique methodological considerations that distinguish eye research from other medical fields.
This article aims to equip clinicians and researchers with practical tools for designing robust studies and selecting appropriate analytical methods, integrating traditional approaches with contemporary innovations. We will explore the continuum from observational to experimental designs, emphasising how research questions drive design choices. Special attention is given to challenges specific to eye research, including the statistical handling of bilateral organ data, the selection of clinically meaningful endpoints, and strategies for minimising bias in studies where traditional masking is impossible. Critically, we also address the ethical and scientific imperative of situating new trials within the context of existing evidence, the transformative potential of platform trials and adaptive designs, and the emerging role of decentralised trial elements in expanding access to clinical research.
Material and Methods
The methods used in this article have been described in Part 1 of this series.1 In brief, this article synthesises evidence from multiple sources to provide comprehensive guidance on clinical research design in eye and vision research. A narrative literature review was conducted using PubMed, Scopus, Web of Science, and the Cochrane Database of Systematic Reviews, covering publications from January 2019 to December 2025, with particular emphasis on methodological innovations published from 2019 onwards, as this period saw the finalisation of ICH E9(R1) on estimands (2019), updated CONSORT guidance (2025), and substantial advances in platform trial and adaptive design methodology. Search terms included combinations of clinical trial design, observational study, ophthalmology, optometry, statistical methods, inter-eye correlation, sample size, bias control, adaptive trials, platform trials, estimands, and decentralised trials.
Inclusion criteria encompassed methodological papers addressing study design in eye research, guidelines from regulatory bodies and professional organisations, systematic reviews of ophthalmic trials, landmark studies demonstrating exemplary design principles, and consensus statements on trial methodology. International regulatory guidance was reviewed including ICH E9(R1) on estimands, FDA guidance on adaptive designs and Bayesian methods, and EMA recommendations on decentralised trials. We excluded non-English publications, conference abstracts without full text, and studies focused solely on basic science research without clinical applications.
The ethical and scientific foundation: Systematic Reviews before new trials
Before designing any new clinical study, researchers have both an ethical and scientific obligation to review existing evidence systematically. This principle, though seemingly obvious, remains inadequately implemented across medical research, contributing substantially to the estimated 85 % of research investment that is wasted globally.7 The 2014 Lancet series on increasing value and reducing waste in research established that research funders and regulators should demand that proposals be justified by systematic reviews of existing evidence.8 This requirement serves multiple purposes: it ensures that new research addresses genuine knowledge gaps, enables sample size calculations to be based on realistic effect estimates, and prevents unnecessary exposure of participants to inferior treatments when evidence already exists.
The consequences of ignoring existing evidence can be profound. A landmark cumulative meta-analysis by Lau et al. demonstrated that the mortality benefit of intravenous streptokinase for myocardial infarction could have been established by 1973, yet over a decade of additional trials enrolled patients to receive a placebo before the treatment became standard practice.9 Similar delays likely occur in eye and vision research, though comprehensive cumulative meta-analyses are less common in our field. The principle that clinical trials should begin and end with systematic reviews reflects both the ethical imperative to minimise unnecessary research and the scientific value of situating new findings within the totality of evidence.10
Recent analysis demonstrates that despite growing awareness, implementation remains inadequate. Jia et al. examined trends in RCT reports citing prior systematic reviews from 2007 to 2021 and found that approximately 40 % of RCT reports still fail to cite any systematic review, though this represents improvement from 65 % a decade earlier.11 In eye and vision research, the situation may be even more concerning, given the relatively young tradition of systematic evidence in the field. Practical implementation requires that researchers search for existing systematic reviews in their area before finalising protocols, conduct new systematic reviews when existing ones are outdated or unavailable, use quantitative synthesis to inform sample size calculations and choice of outcomes, and update systematic reviews to incorporate their new findings upon completion.
For eye and vision research specifically, researchers should consult the Cochrane Eyes and Vision Group reviews, search databases for existing meta-analyses of relevant interventions, and consider whether their proposed study addresses a genuine gap in evidence. When systematic reviews suggest equipoise truly exists, they provide the ethical justification for randomisation. When they reveal that evidence already favours one intervention, researchers must carefully consider whether additional trials are justified and, if so, what specific questions remain unanswered. In some circumstances, proceeding without a formal systematic review may be justified: when investigating entirely novel interventions or technologies with no comparable evidence (e.g., first-in-human trials of gene therapies for inherited retinal diseases), when addressing research questions in previously unstudied populations or settings, or when time-sensitive public health emergencies require rapid evidence generation. In such cases, researchers should document their rationale and conduct the most comprehensive literature search feasible within constraints.
Fundamentals of study design in eye research
The research question: foundation of design selection
Every successful clinical study begins with a clearly formulated research question. The quality and precision of this question determine not only the study design but also the eligibility criteria, sample size, endpoints, and analytical approach. Several frameworks assist researchers in developing answerable, clinically meaningful questions suitable for primary research, with the choice of framework depending on the type of question being asked, Table 1.
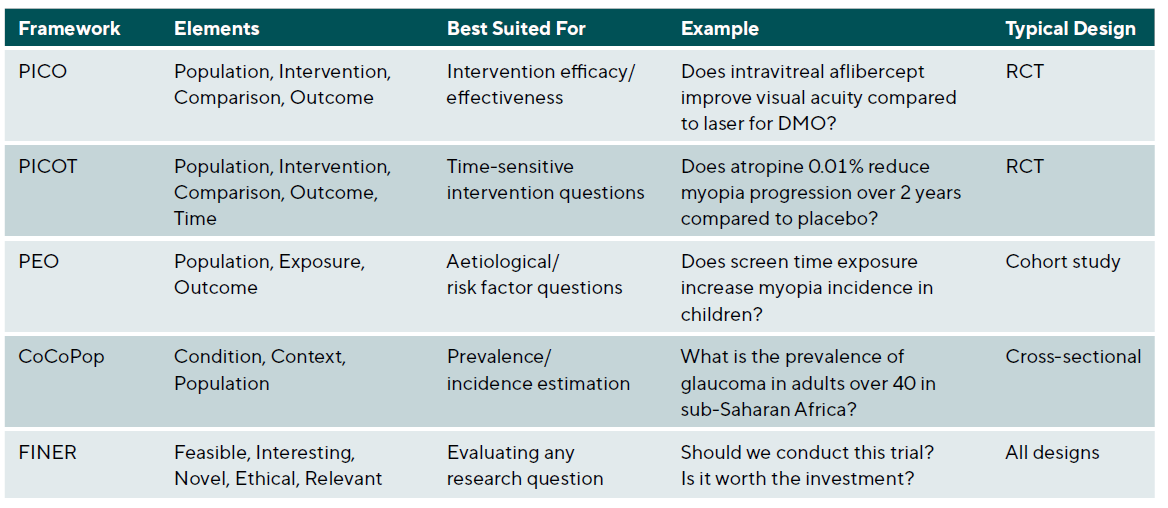
The PEO Framework for aetiological and prognostic studies
Not all clinical research questions involve interventions. When investigating risk factors, prognostic indicators, or disease associations, the PEO framework (Population, Exposure, Outcome) provides more appropriate structure than PICO. This framework, which emerged from epidemiological research traditions, replaces the Intervention and Comparison elements with Exposure, recognising that observational studies examine naturally occurring exposures rather than investigator-assigned treatments.
Consider the aetiological question: “Does high myopia increase the risk of retinal detachment?” Framing this using PEO yields Population (adults with refractive error), Exposure (high myopia, typically defined as spherical equivalent ≤ −6.00 D), and Outcome (retinal detachment). This structure naturally suggests a cohort study design comparing retinal detachment incidence between highly myopic and non-highly myopic individuals, with appropriate adjustment for confounding factors such as age, sex, and history of ocular trauma.
The CoCoPop framework for prevalence and burden studies
Questions about disease frequency require yet another framework. The CoCoPop approach (Condition, Context, Population) structures prevalence and incidence questions where no exposure or intervention comparison exists. Developed within the Joanna Briggs Institute methodology, this framework helps researchers formulate precise descriptive questions.
For example, the question “What is the prevalence of diabetic retinopathy among Type 2 diabetics in urban primary care settings?” becomes Condition (diabetic retinopathy), Context (urban primary care settings), and Population (adults with Type 2 diabetes). This structure clarifies that a cross-sectional design with population-based sampling would be appropriate, and highlights the need for standardised diagnostic criteria and representative sampling within the specified context.
The FINER criteria for research question evaluation
Before committing resources to a clinical trial, researchers should evaluate their question against the FINER criteria.13 These criteria assess whether a research question merits the investment of time, funding, and participant involvement that clinical trials require.
Feasibility asks whether the study can realistically be completed. Can sufficient participants be recruited within a reasonable timeframe? Is the required expertise available? Are the measurements technically achievable? Is funding adequate? For ophthalmic trials, feasibility considerations include access to specialised equipment (OCT, visual field analysers, fundus cameras), availability of qualified outcome assessors, and the challenge of recruiting patients with specific disease stages.
Interest considers whether the question matters to the scientific community and will engage investigators sufficiently to complete a demanding study. A question that fails to generate enthusiasm among potential collaborators will struggle to achieve recruitment targets and maintain quality throughout follow-up.
Novelty asks whether the study will contribute new knowledge. This criterion connects directly to the ethical imperative discussed earlier: systematic review of existing evidence should precede new trials to ensure genuine knowledge gaps exist. A trial that merely replicates well-established findings may waste resources and exposes participants to unnecessary risk.
Ethics requires that genuine equipoise exists regarding the treatments being compared and that the risks to participants are acceptable given potential benefits. In eye and vision research, where outcomes like vision loss can be irreversible, ethical considerations carry particular weight. Sham injection controls, while valuable for masking, require careful ethical justification.
Relevance asks whether the findings will influence clinical practice or health policy. A statistically significant difference that lacks clinical meaningfulness fails this criterion. In ophthalmic trials, clinical relevance often requires defining minimally important differences for outcomes like visual acuity (commonly 5-10 ETDRS letters) or intraocular pressure (typically 2-3 mmHg).
Research question typology: Superiority, non-inferiority, and equivalence
The type of comparison intended fundamentally shapes trial design, sample size calculation, and statistical analysis. Researchers must explicitly specify whether they are asking a superiority, non-inferiority, or equivalence question, as each has distinct design implications.14
Superiority questions ask whether one treatment is better than another and represent the traditional hypothesis-testing framework. The null hypothesis states that treatments are equal, and the trial seeks evidence to reject this in favour of one treatment being superior. Sample size calculations for superiority trials depend on the minimum clinically important difference the investigators wish to detect, the expected variability, and the desired power.
Non-inferiority questions ask whether a new treatment is not unacceptably worse than an established standard. These questions arise when a new treatment offers advantages in cost, convenience, safety, or administration, even if efficacy is not superior. The null hypothesis states that the new treatment is inferior by more than a pre-specified margin, and the trial seeks to reject this hypothesis. Non-inferiority trials require careful specification of the non-inferiority margin, which should be smaller than the established treatment’s effect versus placebo to ensure the new treatment remains superior to no treatment.
In eye and vision research, non-inferiority designs are increasingly relevant. A trial comparing a biosimilar anti-VEGF agent to the reference product might use a non-inferiority design, accepting equivalent efficacy while potentially offering cost advantages. Similarly, comparing a less invasive glaucoma procedure to trabeculectomy might employ non-inferiority testing, hypothesising that the new procedure provides similar IOP control with improved safety.
Equivalence questions ask whether two treatments produce essentially the same outcomes and require demonstrating that any difference falls within a pre-specified equivalence margin in both directions. These designs are less common in clinical practice but arise in bioequivalence studies and some comparative effectiveness contexts.
The distinction between these question types has profound implications for analysis and interpretation. Intention-to-treat analysis, which preserves the benefits of randomisation by analysing participants as randomised regardless of adherence, is conservative for superiority trials (biasing toward the null) but anti-conservative for non-inferiority trials (biasing toward equivalence). Consequently, non-inferiority trials should report both intention-to-treat and per-protocol analyses, with both required to support a non-inferiority conclusion.
Hierarchy of evidence and study types
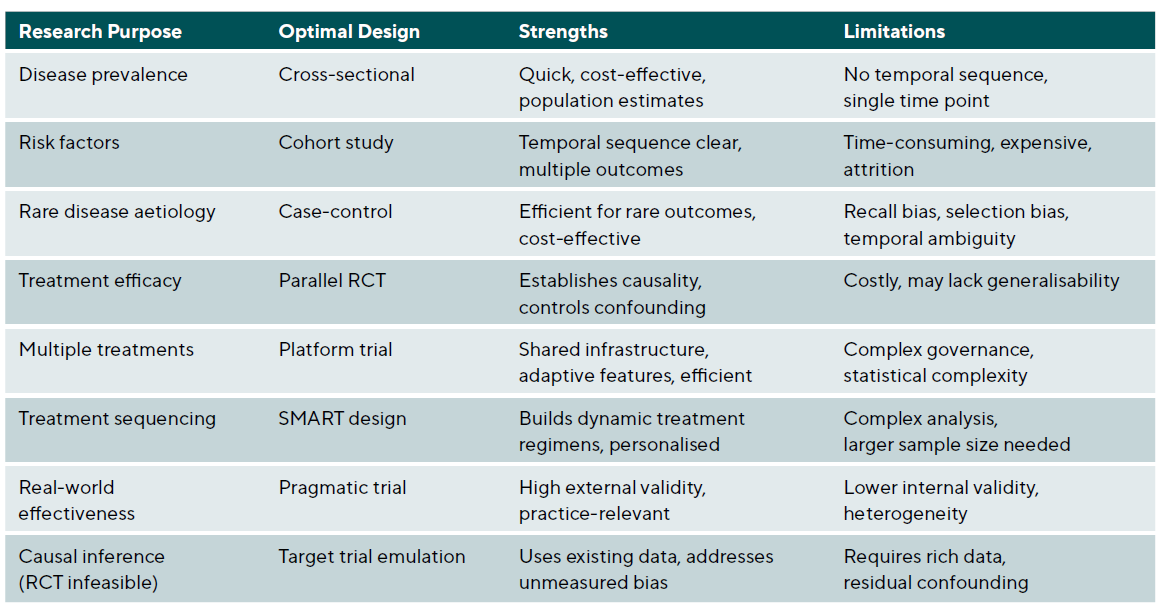
The evidence pyramid traditionally places systematic reviews and meta-analyses at the apex, followed by randomised controlled trials, cohort studies, case-control studies, and case series. However, this hierarchy assumes that the research question involves treatment efficacy and that traditional frequentist RCTs are the only option. Different questions require different optimal designs, and modern trial methodology offers alternatives that may be more efficient or ethical for specific contexts. Critically, the rigid hierarchy has been increasingly challenged as oversimplistic.15,16 RCTs may be infeasible for rare diseases, unethical when effective treatments exist, or impractical for long-term outcomes such as cataract development over decades. In such circumstances, well-conducted observational studies with appropriate confounding control may provide the best available evidence. The key principle is matching the study design to the research question rather than reflexively privileging one design type. See Table 2 and Table 3.
Observational studies versus experimentation
The most important division in clinical research methodology lies between observational and experimental studies. This distinction rests on a single question: does the investigator assign the exposure or intervention?
In observational research, the investigator examines exposures that occur naturally, such as smoking habits, dietary patterns, genetic variants, environmental conditions, or treatment choices made by clinicians and patients. Because these exposures are not randomly assigned, they may be associated with other factors (confounders) that independently affect the outcome, making causal inference challenging.
In experimental research, the investigator assigns the intervention, ideally using randomisation to ensure that treatment groups are comparable at baseline. This manipulation breaks the link between treatment assignment and potential confounders, enabling more confident causal conclusions. However, experimental designs are not always feasible or ethical, necessitating observational approaches for many important questions.

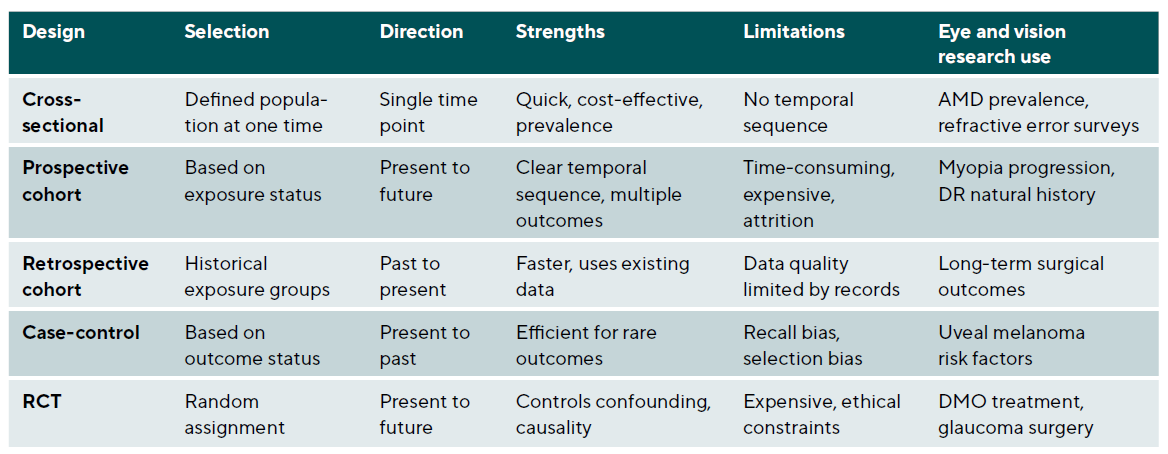
Temporal direction in research
Studies vary in their temporal direction, timing of data collection, and analytical approach. Three primary designs - cross-sectional, cohort, and case-control studies – form the foundation of observational research. Landmark observational studies in eye and vision research have employed these designs to generate foundational knowledge: the Blue Mountains Eye Study, a population-based cohort study from Australia, has contributed substantially to our understanding of age-related macular degeneration risk factors and natural history 3; the Collaborative Longitudinal Evaluation of Ethnicity and Refractive Error (CLEERE) Study has provided critical prospective data on myopia development in children 4; and the Rotterdam Study has informed our understanding of genetic and environmental factors in eye disease.5

Cross-sectional studies
Cross-sectional studies provide a snapshot of disease prevalence and associated factors at a single time point. In eye and vision research, these studies have been instrumental in establishing the global burden of visual impairment and identifying populations at risk. Design considerations include sampling strategy, where population-based sampling provides the most generalisable results but requires substantial resources, while clinic-based sampling is more feasible but may introduce selection bias. Standardised measurements using validated instruments and standardised protocols ensure comparability across sites and studies; the Early Treatment Diabetic Retinopathy Study (ETDRS) chart has become the gold standard for visual acuity assessment. Quality control through training and certifying examiners, implementing duplicate grading for imaging, and monitoring inter-rater reliability is essential for data quality.
Statistical analysis of cross-sectional data requires prevalence estimates with confidence intervals accounting for the sampling design. When examining associations, logistic regression for binary outcomes or linear regression for continuous outcomes are commonly used, with adjustment for potential confounders. Complex survey designs require appropriate weighting and variance estimation methods.
Cohort Studies
Cohort studies follow individuals over time to assess the development of outcomes, making them ideal for studying disease incidence and identifying risk factors. Prospective cohort studies are particularly valuable for understanding the natural history of eye diseases. Key design elements include cohort selection with clear inclusion and exclusion criteria, considering whether to use an inception cohort enrolled at disease onset or a prevalent cohort enrolled at various disease stages. Follow-up protocols should establish regular assessment intervals based on expected disease progression rates; more frequent assessments capture changes better but increase costs and participant burden. Retention strategies minimising attrition through participant engagement, flexible scheduling, and travel reimbursement are essential, and investigators should plan for 20 - 30 % loss to follow-up in sample size calculations.
Statistical considerations for cohort data include survival analysis methods like Kaplan-Meier curves and Cox proportional hazards models that account for varying follow-up times. For repeated measurements, mixed-effects models or generalised estimating equations handle within-subject correlation appropriately.
Case-Control studies
Case-control studies are particularly efficient for studying rare eye diseases or outcomes with long latency periods. By starting with the outcome and looking backwards for exposures, they require smaller sample sizes than cohort studies. Critical design decisions include case definition using strict diagnostic criteria to ensure cases truly have the condition of interest and considering whether to include incident or prevalent cases. Control selection requires that controls come from the same source population as cases and be at risk for developing the outcome; hospital controls may share healthcare-seeking behaviour with cases but might have other conditions affecting exposures. Matching on key confounders like age and sex can improve efficiency but requires matched analysis and prevents studying matched factors
as risk factors.

Why randomisation matters: The problem of confounding
The hierarchy of evidence places randomised controlled trials above observational studies primarily because randomisation addresses the fundamental problem of confounding. A confounder is a variable that is associated with both the exposure and the outcome, potentially creating a spurious association or masking a true one.

When observational designs are necessary or preferred
Despite the advantages of randomisation, observational studies remain essential in clinical research for several reasons. First, many important questions cannot be addressed experimentally - we cannot randomise individuals to smoking or high myopia to study their effects on disease risk. Second, randomisation may be unethical when evidence already favours one treatment or when the intervention carries substantial risk. Third, observational studies reflect real-world practice, capturing effectiveness in routine clinical care rather than efficacy under controlled conditions. Fourth, observational designs are often more feasible for rare diseases, long-term outcomes, or hypothesis generation.
Non-Randomised Controlled Trials and causal inference
Non-randomised controlled trials, also known as quasi-experimental studies, evaluate interventions without random allocation. These designs are valuable when randomisation is impractical, unethical, or when evaluating health system interventions implemented at the population level. Modern methodological advances have substantially strengthened the causal inferences possible from non-randomised data.
Before-After studies and interrupted time series
Before-after studies compare outcomes before and after an intervention within the same population. In eye and vision research, these are commonly used to evaluate quality improvement initiatives or the impact of new technologies. While feasible and pragmatic, these studies are vulnerable to temporal trends, regression to the mean, and the Hawthorne effect. Including a control group that does not receive the intervention strengthens causal inference.

Interrupted time series designs assess the impact of an intervention by analysing trends before and after implementation. Multiple pre- and post-intervention measurements help distinguish intervention effects from underlying trends. The analytical approach uses segmented regression analysis to model level change and slope change at the intervention point. Autoregressive integrated moving average (ARIMA) models can account for autocorrelation in the time series data, providing more valid standard errors and confidence intervals.
Target trial emulation
When randomised trials are infeasible, target trial emulation offers a rigorous framework for causal inference from observational data. Developed by Hernán and Robins, this methodology specifies that observational analyses should explicitly emulate a hypothetical target trial by defining seven protocol components: eligibility criteria, treatment strategies, assignment procedures, follow-up period, outcome, causal contrast of interest, and analysis plan.20 By designing observational analyses to mirror a well-designed RCT, researchers can identify and avoid common biases that afflict naive observational comparisons. In ophthalmology, target trial emulation could be applied to questions such as comparing anti-VEGF treatment regimens using electronic health record data, evaluating the long-term safety of refractive surgery using registry data, or assessing the effectiveness of different glaucoma treatment sequences in populations underrepresented in clinical trials.
The framework is particularly valuable for avoiding immortal time bias, a common error in observational studies where the period between cohort entry and treatment initiation is incorrectly attributed to the treatment group, artificially improving outcomes. Hernán et al. demonstrated how to apply the framework systematically, with subsequent regulatory acceptance of real-world evidence for certain decisions under the 21st Century Cures Act.21 However, Concato and Corrigan-Curay emphasise that simply labelling data as ‘real-world’ does not specify study architecture or data quality; the rigour of the analytical approach determines validity.22
Propensity Score methods
Propensity score methods attempt to balance confounders between treated and untreated groups in observational data, mimicking randomisation. The propensity score represents the probability of receiving treatment given observed covariates. Implementation strategies include matching, which pairs treated and untreated subjects with similar propensity scores; stratification, which divides subjects into strata based on propensity score quintiles; weighting, which uses inverse probability of treatment weighting in the analysis; and adjustment, which includes the propensity score as a covariate in regression models. Each approach has advantages and limitations, and the choice depends on the specific research context and data structure.
Randomised Controlled Trials in eye care
Randomised controlled trials remain the gold standard for evaluating treatment efficacy, as randomisation balances both known and unknown confounders between groups. The rigour of RCTs has established many cornerstones of modern eye care, from anti-VEGF therapy for retinal diseases to surgical techniques for glaucoma. However, the traditional two-arm parallel RCT represents just one option within a diverse landscape of experimental designs, and modern methodology offers numerous refinements and alternatives.
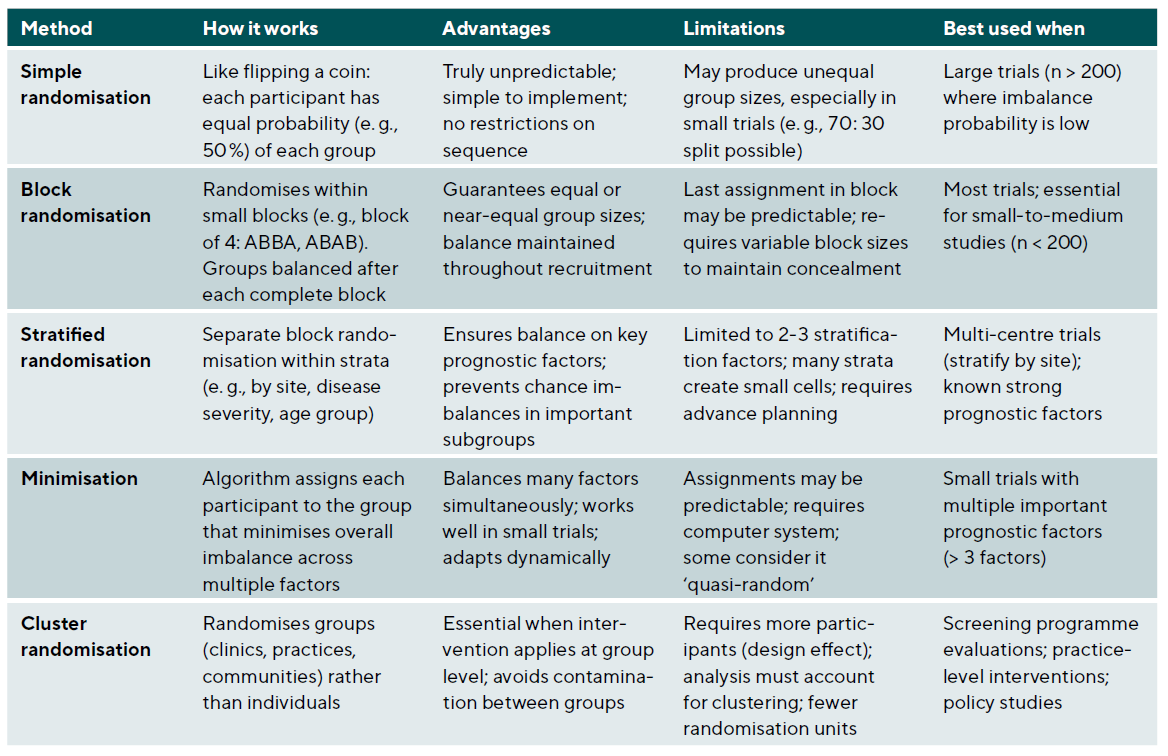
Randomisation methods
The randomisation method should ensure unpredictability while achieving balance between groups, Table 4. Simple randomisation gives each participant an equal probability of assignment to any group and works well for large samples but may result in imbalanced groups in smaller studies. Block randomisation ensures balance by randomising within blocks of predetermined size; for example, blocks of four with two treatments ensure equal numbers after every fourth participant. Stratified randomisation performs separate randomisation within strata of important prognostic factors, ensuring balance across these factors. Minimisation dynamically assigns participants to minimise imbalance across multiple factors simultaneously and is particularly useful for smaller trials with multiple important covariates.
Allocation concealment and blinding
Allocation concealment prevents selection bias by ensuring that investigators cannot predict or influence group assignment. Methods include central randomisation systems (web-based or telephone), sequentially numbered opaque sealed envelopes, and pharmacy-controlled randomisation for drug trials. Blinding (masking) prevents performance and detection bias, but eye and vision research presents unique challenges. For surgical interventions, true sham surgery is often unethical, necessitating masked outcome assessors and standardised post-operative care protocols. For device studies, different device appearances may preclude patient blinding, so efforts should focus on blinding outcome assessors and data analysts. For injection studies, sham injections can maintain blinding but require ethical justification; masked visual acuity examiners and reading centres for imaging outcomes provide alternative approaches.
Sample size calculation
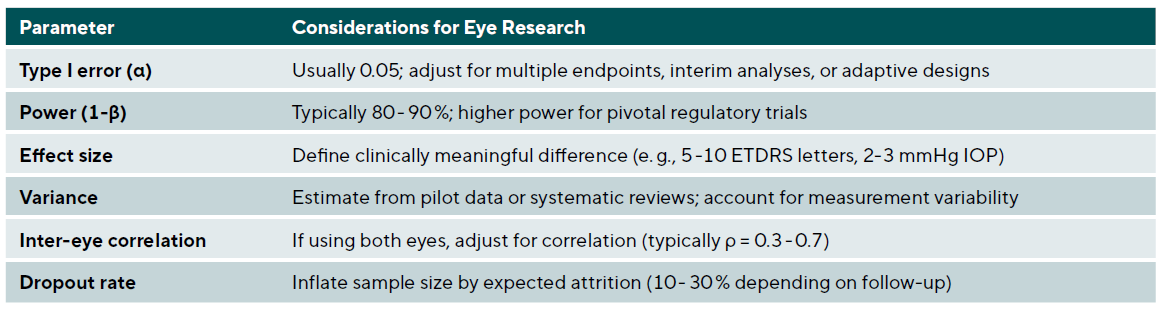
Adequate sample size ensures sufficient power to detect clinically meaningful differences while avoiding waste of resources. Key parameters include Type I error (α), usually set at 0.05 with consideration of adjustment for multiple endpoints or interim analyses; power (1-β), typically 80 - 90 % with higher power for pivotal trials; effect size, defined as the clinically meaningful difference (e.g., 5 ETDRS letters for visual acuity); variance estimated from pilot studies or literature; inter-eye correlation if using both eyes; and anticipated dropout rate to inflate sample size appropriately.
For studies using both eyes, the effective sample size is reduced due to correlation. The design effect can be calculated as: Design Effect = 1 + (m − 1) × ρ, where m is the number of observations per subject (2 for both eyes), and ρ is the inter-eye correlation coefficient, typically ranging from 0.3 to 0.7 for many ophthalmic outcomes. For example, if a study initially calculates that 100 subjects (200 eyes) are needed assuming independence, and the true inter-eye correlation is ρ = 0.5, the design effect = 1 + (2 − 1) × 0.5 = 1.5. The effective sample size is therefore 200 / 1.5 = 133 independent units, requiring approximately 150 subjects (300 eyes) to achieve the originally planned power. Table 5 summarises key parameters for sample size calculation in ophthalmic trials.

The Estimands Framework: Defining what trials measure
The ICH E9(R1) addendum, effective since 2020, introduced the estimands framework – a structured approach to defining precisely what treatment effect a trial intends to estimate.23 This framework represents a fundamental shift in how clinical trials are designed, conducted, and analysed, ensuring that the research question, trial design, and statistical analysis are explicitly aligned. For eye and vision research, where treatment discontinuation, rescue therapy, and switching are common, the estimands framework has particular relevance.
The five attributes of an Estimand
An estimand has five defining attributes that together specify exactly what treatment effect is being estimated.
1. The treatment attribute specifies the intervention being evaluated, including dose, duration, and any relevant aspects of administration.
2. The population attribute defines who the treatment effect applies to, including inclusion and exclusion criteria and any restrictions on generalisability.
3. The variable attribute identifies the endpoint or outcome of interest, including how and when it is measured.
4. The intercurrent events attribute addresses how post-randomisation events that affect interpretation will be handled.
5. Finally, the population-level summary attribute specifies the statistical measure of effect, such as difference in means, hazard ratio, or odds ratio.
The concept of intercurrent events is central to the framework. These are post-randomisation occurrences that affect either the interpretation or the existence of outcomes, including treatment discontinuation, rescue medication use, treatment switching, and death. Rather than treating these as missing data problems to be handled in the analysis phase, the estimands framework requires explicit decisions about how each type of intercurrent event relates to the scientific question of interest.
Strategies for handling intercurrent events
Five strategies exist for handling intercurrent events, each answering a different clinical question.
1. The treatment policy strategy includes all outcomes regardless of intercurrent events, essentially answering ‘What happens if we assign this treatment?’ regardless of what patients actually do. This approach aligns with traditional intention-to-treat analysis.
2. The hypothetical strategy estimates the treatment effect that would have been observed if the intercurrent event had not occurred, answering questions like ‘What would the effect be if patients had not discontinued treatment?’
3. The composite strategy incorporates the intercurrent event into the endpoint definition, such as defining the outcome as ‘vision improvement without requiring rescue therapy.’
4. The while-on-treatment strategy includes only outcomes while on the randomised treatment, useful when the effect of actual treatment receipt is of interest.
5. The principal stratum strategy focuses on subsets of patients defined by their potential intercurrent event behaviour, such as ‘patients who would adhere to treatment regardless of assignment.’
Application to eye and vision research
The practical implications for ophthalmic trials are substantial. Kahan et al. provided an accessible primer using worked examples that clinicians can adapt 24, while Ratitch et al. offered detailed guidance on defining efficacy estimands.25 Consider a trial of a new anti-VEGF agent for diabetic macular oedema where patients may discontinue the study drug and receive rescue treatment with an alternative anti-VEGF agent. Different estimands answer different questions:
• A treatment policy estimand asks about the effect of initiating the new agent versus comparator regardless of subsequent treatment changes, which may be most relevant for health policy decisions.
• A hypothetical estimand asks what the effect would be if no one received rescue therapy, though this may be unrealistic clinically.
• A composite estimand might define success as visual improvement without requiring rescue therapy, capturing both efficacy and tolerability.
In glaucoma trials, where medication discontinuation and switching are common, the estimands framework helps clarify whether the trial aims to assess the effect of the initial treatment assignment (treatment policy), the effect if patients had maintained their assigned treatment (hypothetical), or the effect while actually receiving the assigned medication (while-on-treatment). Each answers a legitimate but distinct clinical question, and the choice should be driven by the intended use of the evidence. Fletcher and colleagues documented implementation experience marking two years of applying the new framework, providing practical guidance for researchers transitioning to estimand-based trial design.26
Advanced trial designs
The traditional two-arm parallel RCT, while rigorous, is not always the most efficient or informative design for addressing clinical questions. Modern trial methodology offers sophisticated alternatives that can improve efficiency, enable adaptive learning, and better address the complexity of real-world treatment decisions.
Crossover and Factorial designs
Crossover trials have each participant serve as their own control by receiving all treatments in sequence. This design is particularly powerful for chronic conditions with reversible treatment effects, offering reduced sample size requirements (typically 25 - 50 % of parallel design), control for between-subject variability, and particular suitability for comparing eye drops or contact lenses. However, limitations include the requirement for adequate washout periods to avoid carryover effects, unsuitability for curative treatments or progressive diseases, and potential period effects if disease state changes over time.
Factorial designs test multiple interventions simultaneously, allowing assessment of individual effects and interactions. A 2 × 2 factorial design tests two interventions, each at two levels. For example, testing both a new surgical technique and post-operative medication in cataract surgery creates four groups: standard surgery plus placebo, standard surgery plus medication, new surgery plus placebo, and new surgery plus medication. This design efficiently evaluates two questions within a single trial while also assessing whether the interventions interact.
Platform trials and master protocols
The COVID-19 pandemic demonstrated the extraordinary power of platform trials - perpetual infrastructure that can evaluate multiple interventions simultaneously against shared control arms. The RECOVERY trial, which enrolled over 45,000 patients across 176 UK NHS hospitals, established dexamethasone as the first treatment to reduce COVID-19 mortality, fundamentally changing practice within months of initiation.27 Woodcock and LaVange’s seminal paper from the FDA established the taxonomy distinguishing platform, basket, and umbrella trials under the ‘master protocol’ framework.28
Platform trials offer several key advantages over traditional RCTs: shared infrastructure reduces per-comparison costs; adaptive features allow ineffective treatments to be dropped while adding new treatments as they become available; Bayesian methods enable continuous learning and more intuitive probability statements; and shared control arms improve efficiency while maintaining scientific rigour. The Adaptive Platform Trials Coalition provided comprehensive recommendations for design, conduct, and reporting 29, while Berry et al. articulated the efficiency arguments favouring platform approaches.30
For eye and vision research, platform trials offer particular promise for conditions where multiple treatments are under investigation. A platform trial for diabetic macular oedema could simultaneously evaluate multiple anti-VEGF agents, different dosing regimens, and combination approaches against a shared standard-of-care arm. Similarly, platform infrastructure for glaucoma device trials could efficiently compare multiple minimally invasive glaucoma surgery devices while new technologies continue entering the market. Park et al.’s systematic review identified 83 master protocol trials through 2019, demonstrating growing adoption of these approaches.31
Sequential Multiple Assignment Randomised Trials (SMARTs)
Sequential Multiple Assignment Randomised Trials address a fundamental clinical reality: optimal treatment often requires adaptive sequencing based on patient response. Unlike traditional RCTs that test fixed interventions, SMARTs randomise patients at multiple decision points to build evidence for dynamic treatment regimens. Susan Murphy’s foundational 2005 paper introduced the theoretical framework, demonstrating that sequential randomisation is necessary because past treatment effects may have delayed consequences that influence subsequent treatment choices.32
The methodology has since been refined through accessible tutorials by Almirall and Nahum-Shani, who provided practical guidance on design, power analysis, and longitudinal data analysis.33,34 SMARTs are particularly appropriate for chronic disease management where treatment must adapt over time. In eye and vision research, myopia progression management presents an ideal application: children might be initially randomised to atropine versus orthokeratology, then re-randomised based on response to continue current treatment versus switch or augment with the alternative approach. Artman et al. addressed sample size considerations specifically for comparing embedded dynamic treatment regimens 35, while Nahum-Shani et al. provided tutorials for longitudinal analysis of SMART data.36
Bayesian adaptive designs
Bayesian methods provide clinicians with the probability that a treatment works given observed data – a more intuitive interpretation than frequentist p-values. The framework incorporates prior information, updates beliefs as data accumulate, and enables predictive probability calculations for futility and success. The FDA’s 2010 guidance on the use of Bayesian statistics in medical device clinical trials remains the foundational regulatory document for these methods, with subsequent international methodological reinforcement provided through ICH E9(R1), underscoring their relevance for ophthalmic device evaluation.23,37
The key insight is that ‘today’s posterior becomes tomorrow’s prior’ - Bayesian learning is inherently sequential. This enables efficient interim analyses without the rigid stopping rules of frequentist approaches. Rather than asking whether we can reject the null hypothesis at a predetermined alpha level, Bayesian analyses ask what probability we should assign to meaningful treatment effects given all accumulated evidence. Muehlemann and colleagues’ 2023 tutorial specifically addresses the knowledge gap that limits Bayesian adoption, explaining concepts starting from familiar clinical practice and using the Pfizer/BioNTech COVID-19 vaccine trial as a detailed case study.38
Adaptive trials allow pre-specified modifications based on accumulating data without compromising validity. Common adaptations include sample size re-estimation based on observed variance, dropping ineffective treatment arms, enrichment strategies focusing on responsive subgroups, and seamless phase II/III designs. For rare eye diseases where traditional trials would require impractically long enrolment periods, Bayesian adaptive approaches enable efficient evidence generation with smaller sample sizes while maintaining appropriate uncertainty quantification.
When choosing between frequentist and Bayesian approaches, clinicians should consider several practical factors. Frequentist methods offer familiarity, regulatory precedent, and straightforward interpretation of confidence intervals, making them suitable when prior information is limited or controversial. Bayesian methods excel when incorporating prior knowledge is appropriate (e.g., from earlier trial phases), when continuous monitoring with flexible stopping is desired, when sample sizes are small (as in rare eye diseases), or when probability statements about treatment effects are more clinically intuitive than hypothesis testing. The choice is not mutually exclusive: many modern trials use Bayesian methods for interim decisions while reporting frequentist statistics for regulatory submissions.
Decentralised clinical trials
COVID-19 forced rapid adoption of decentralised clinical trial (DCT) elements, with over 500 DCTs conducted annually since 2020. These trials replace or supplement traditional site visits with remote data collection via telehealth, wearables, electronic patient-reported outcomes, and home nursing visits. The FDA’s September 2024 final guidance and EMA’s 2022 recommendations establish regulatory frameworks emphasising patient safety and data integrity.39,40 Park et al. provided a comprehensive landscape analysis of DCT development and regulatory guidance.41
Electronic diaries and electronic clinical outcome assessments show highest current adoption (51 - 56 % across major pharmaceutical companies), while local physician networks and telemedicine show the greatest growth potential.42 Digital endpoints require rigorous ‘fit-for-purpose’ validation covering technological validity and clinical validation including repeatability, patient/control differentiation, and correlation with traditional endpoints. Kruizinga et al. developed a structured framework for validating novel digital endpoints.43
For eye and vision research, DCT elements present both opportunities and limitations. Home visual acuity testing using validated smartphone applications shows promise for monitoring trials, as do remote fundus photography systems and visual function questionnaires administered electronically. Patient-reported outcome measures for symptoms and quality of life are particularly amenable to remote collection. However, core ophthalmic assessments including slit-lamp examination, Goldmann applanation tonometry, optical coherence tomography, and automated perimetry remain fundamentally site-based due to equipment requirements and the need for trained operators.
Hybrid designs combining DCT elements with essential site visits appear optimal for most ophthalmic research. For example, a dry eye trial might conduct screening and periodic safety assessments at sites while collecting daily symptom diaries and quality-of-life measures remotely. This approach expands geographic access, reduces participant burden, and enables more frequent outcome assessment while maintaining the specialised examinations essential for eye care research. The Clinical Trials Transformation Initiative’s foundational recommendations, which predated the pandemic, have shaped subsequent regulatory guidance and provide a framework for implementing DCT elements responsibly.42
Special considerations for eye and vision research
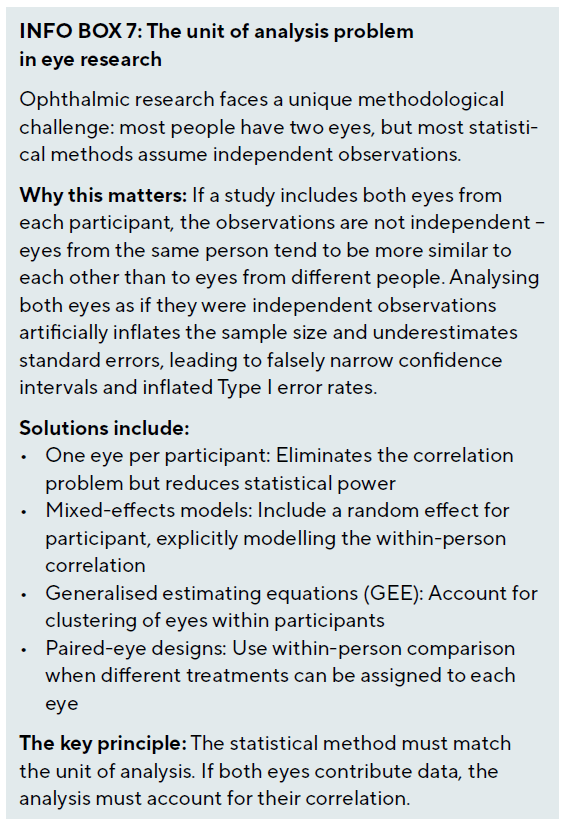
Managing paired organ data
The paired nature of eyes presents unique statistical challenges that distinguish ophthalmic research from most other medical fields. Different approaches are appropriate depending on the research question and study design. Selecting one eye per subject eliminates correlation issues but reduces power; selection methods include random selection, choosing the worse or better eye based on disease severity, or using the first eye to meet eligibility criteria. Including both eyes increases power but requires appropriate statistical methods to account for the correlation: mixed-effects models include random effects for subjects, generalised estimating equations specify correlation structure between eyes, and robust standard errors adjust for clustering within subjects.
Paired eye designs assigning different treatments to each eye of the same patient provide perfect matching but require careful consideration. Systemic effects may contaminate the control eye, introducing bias. Patient preference or psychological effects may differ when both treatments are experienced simultaneously. These designs are most appropriate for localised treatments without systemic absorption, such as comparing two different contact lens designs.
Selecting clinically meaningful endpoints
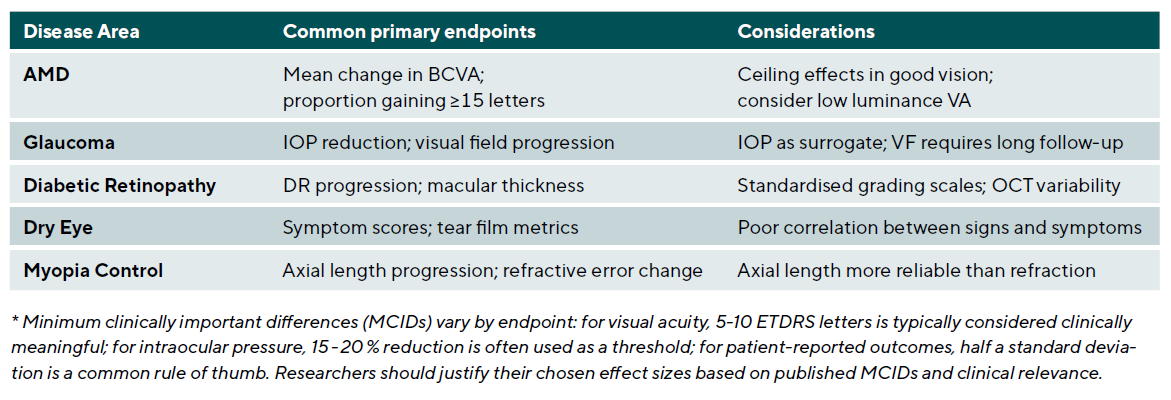
Choosing appropriate primary outcomes is crucial for trial success and clinical relevance. Endpoints should be clinically relevant and meaningful to patients and practitioners, reliable and valid with reproducible measurement properties, sensitive to change and able to detect treatment effects, and feasible to measure in the study setting. Table 6 summarises common primary endpoints across major ophthalmic disease areas.
Pragmatic trials and real-world evidence
Pragmatic trials prioritise external validity and real-world applicability, in contrast to explanatory trials focused on controlled efficacy demonstration. Ford and Norrie provided a comprehensive overview distinguishing these approaches 44, emphasising that the choice between pragmatic and explanatory designs depends on the clinical question being addressed. The PRECIS-2 tool provides a nine-domain ‘wheel’ helping trialists explicitly design trials matching their intended purpose, with each domain scored from 1 (very explanatory) to 5 (very pragmatic).45
The nine PRECIS-2 domains assess eligibility criteria, recruitment path, setting, organisation, flexibility in delivery, flexibility in adherence, follow-up, primary outcome, and primary analysis. By explicitly scoring each domain, researchers can identify whether their design matches their intended use of the results. A highly explanatory trial with strict eligibility, intensive monitoring, and per-protocol analysis may demonstrate whether an intervention can work under ideal conditions, but results may not generalise to routine practice. A pragmatic trial with broad eligibility, usual care comparators, and intention-to-treat analysis addresses whether an intervention does work in practice.
For eye and vision research, pragmatic trials are particularly valuable for comparing surgical techniques where efficacy is established but comparative effectiveness in routine practice is unknown, evaluating screening programmes where implementation factors strongly influence outcomes, and assessing care pathways where the intervention is a system-level change rather than a discrete treatment. Registry-based randomised trials represent an emerging hybrid approach where randomisation is embedded within existing clinical registries, enabling large-scale pragmatic trials with minimal additional infrastructure.

Statistical analysis of clinical research data
Analysis principles
The intention-to-treat (ITT) principle analyses participants as randomised regardless of adherence, preserving randomisation benefits and providing conservative efficacy estimates. Per-protocol analysis includes only participants who completed the protocol as planned, potentially overestimating treatment effects but useful for understanding efficacy under ideal conditions. Best practice involves conducting both analyses, with ITT as the primary analysis for superiority trials. For non-inferiority trials, both ITT and per-protocol analyses should demonstrate non-inferiority, as ITT alone can bias towards equivalence. The estimands framework clarifies how these choices relate to the scientific question of interest.
Missing data are inevitable in clinical trials and can bias results if not handled appropriately. Modern approaches include multiple imputation, which creates multiple plausible values for missing data based on observed patterns; mixed models, which use all available data under missing at random assumptions; and sensitivity analyses, which test robustness under different missing data assumptions including missing not at random scenarios.
Modern multiplicity methods
Clinical trials often involve multiple endpoints, time points, subgroups, and treatment comparisons, increasing the risk of Type I errors. Modern multiplicity methods have advanced far beyond simple Bonferroni correction. Bretz et al. introduced the graphical approach to multiple testing procedures, representing multiple test procedures as directed weighted graphs where nodes correspond to hypotheses.46,47 This visualisation enables clinicians and regulators to understand complex testing hierarchies intuitively.
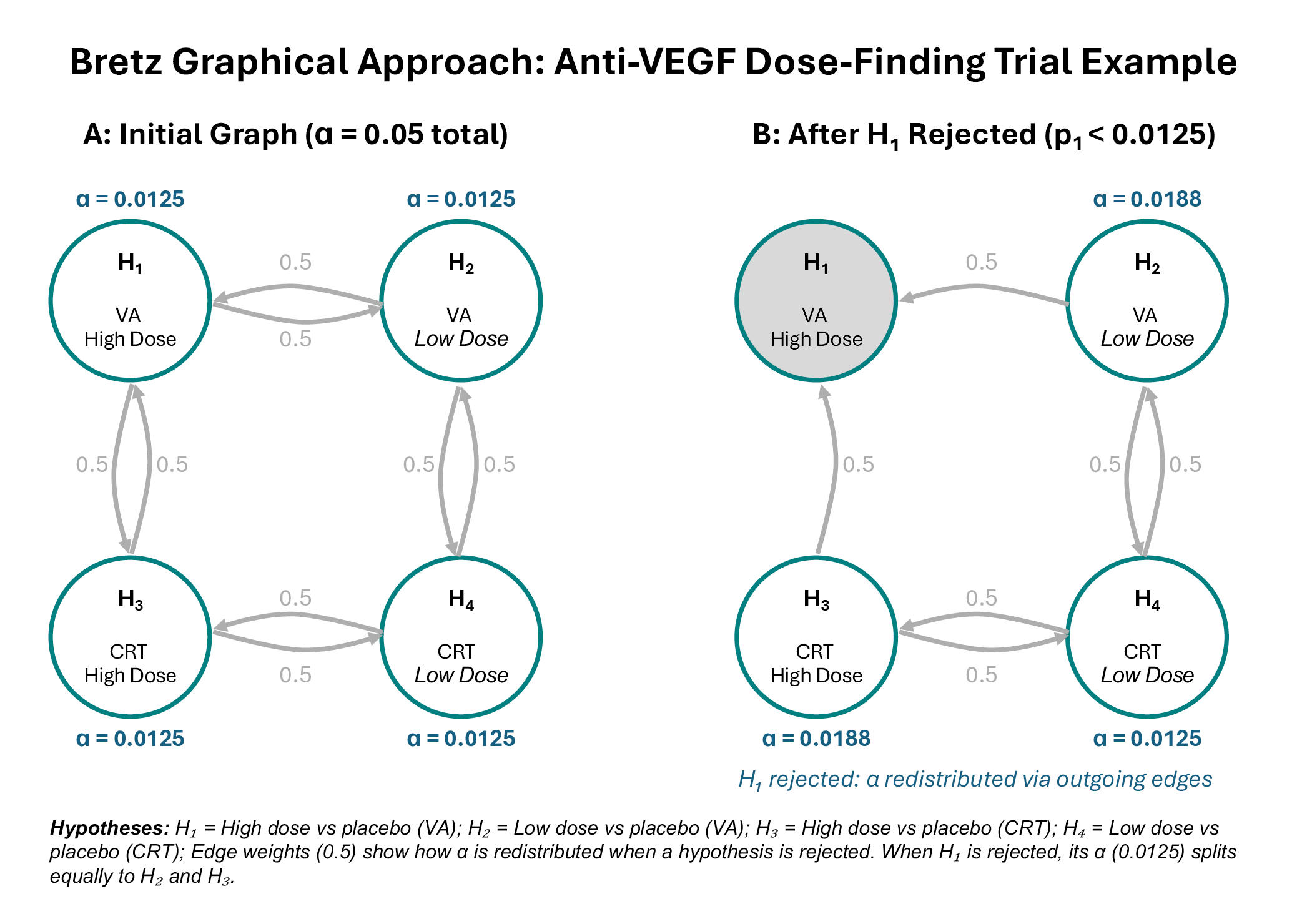
Worked example: Anti-VEGF Dose-Finding Trial (see Figure 1). Consider a phase III trial comparing high-dose and low-dose anti-VEGF against placebo for wet AMD, with co-primary endpoints of visual acuity (VA) and central retinal thickness (CRT). This creates four hypotheses: H₁ (high dose vs placebo, VA), H₂ (low dose vs placebo, VA), H₃ (high dose vs placebo, CRT), and H₄ (low dose vs placebo, CRT).
In the initial graph (Figure 1A), the total α = 0.05 is divided equally among all four hypotheses (α = 0.0125 each). The edge weights of 0.5 indicate that when any hypothesis is rejected, half of its α goes to the hypothesis testing the same dose for the other endpoint, and half goes to the hypothesis testing the other dose for the same endpoint. This structure reflects the clinical logic that evidence for one dose informs our confidence in testing both endpoints for that dose, and that evidence for one endpoint informs testing both doses for that endpoint.
Suppose the trial finds p₁ = 0.008 for the high-dose VA comparison (H₁). Since 0.008 < 0.0125, we reject H₁. The graph updates (Figure 1B): H₁ is removed, and its α = 0.0125 is redistributed according to the outgoing edge weights. H₂ (low dose VA) receives 0.0125 × 0.5 = 0.00625, bringing its total to 0.01875. H₃ (high dose CRT) also receives 0.00625, bringing its total to 0.01875. H₄ remains at 0.0125. The procedure continues with the updated α levels, providing more power to test the remaining hypotheses than would be available under Bonferroni correction.
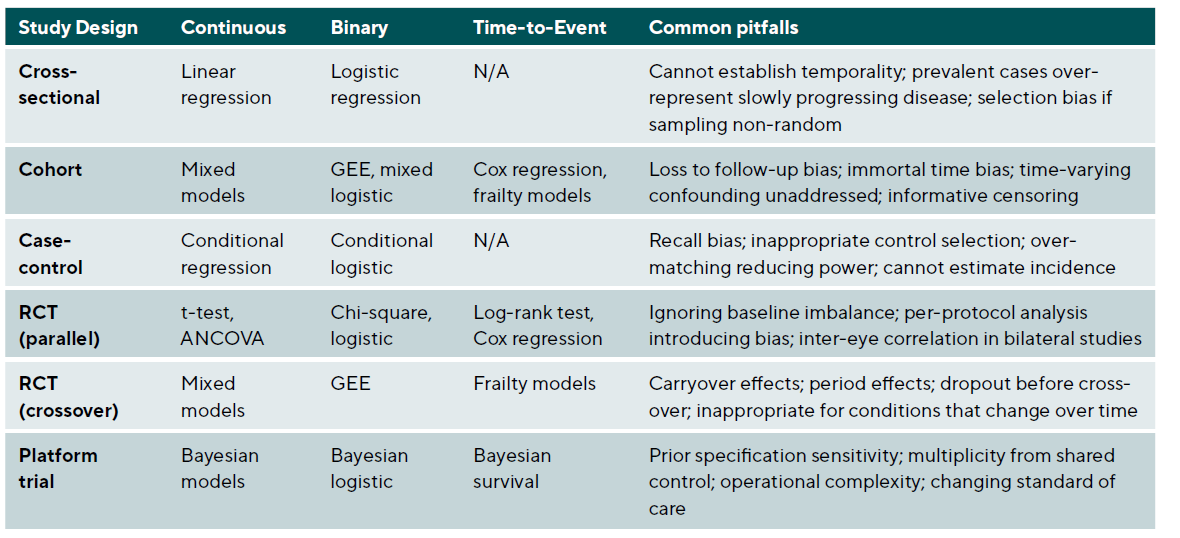
The graphical approach has been extended to incorporate weighted Simes tests and parametric tests that leverage correlation between endpoints for increased power.48 Gatekeeping procedures address trials with primary and secondary hypothesis families, where secondary analyses proceed only after gatekeeper hypotheses are rejected.49 Li et al.’s accessible tutorial uses a question-and-answer format organised by the PICOTA framework, addressing common scenarios including multi-arm trials, multiple outcomes, and subgroup analyses.50 Table 7 summarises appropriate statistical methods for different study designs and outcome types.
Reporting standards and quality assurance
Transparent and complete reporting enables critical appraisal and replication. The past five years have seen substantial updates to reporting guidelines reflecting methodological advances. The CONSORT 2025 statement represents the first major update since 2010, integrating advances from recent extensions and addressing contemporary trial features including adaptive designs and estimands.51 Key guidelines include CONSORT for randomised controlled trials, STROBE for observational studies, TREND for non-randomised evaluations, and STARD for diagnostic accuracy studies.
Adaptive designs require additional reporting elements addressed by the ACE (Adaptive designs CONSORT Extension) statement, a Delphi-developed checklist essential for platform trials.52 SPIRIT-PRO provides 16 items for including patient-reported outcomes in protocols 53, while CONSORT-Outcomes 2022 specifies 17 outcome-specific items including the five core elements of outcome definition.54 These extensions ensure that trials using modern methods are reported with sufficient detail for replication and critical appraisal.
Data sharing transparency became mandatory under ICMJE requirements effective 2018, requiring statements about what data will be shared, when, and through what mechanisms.55 This represents an ethical obligation arising from trial participants’ contributions and enables secondary analyses that can extend the value of trial data. Prospective trial registration (e.g., ClinicalTrials.gov, ISRCTN) prevents selective reporting and publication bias; registration should occur before enrolment begins and include primary outcomes, sample size, and analysis plan.
Case studies: Applying design principles
Case 1: SMART design for myopia progression management
Research question: What is the optimal sequence of myopia control interventions based on initial treatment response in school-aged children?
Design: Sequential Multiple Assignment Randomised Trial with two stages. In Stage 1, children aged 6-12 years with myopia progression ≥ 0.50 D/year are randomised to low-dose atropine (0.01 %) versus orthokeratology. After 6 months, responders (progression slowed to < 0.25 D/year) continue assigned treatment, while non-responders are re-randomised to either add the alternative treatment or switch to combination therapy from the outset.
Estimand specification: The primary estimand uses a treatment policy strategy, comparing embedded dynamic treatment regimens regardless of adherence, answering the question most relevant to clinical practice: ‘If we follow this decision rule, what outcomes can we expect?’
Key considerations: Standardised cycloplegic refraction and axial length measurement protocols ensure consistent outcome assessment. The 6-month response criterion balances early identification of non-responders against sufficient time for treatment effects to manifest. Sample size calculations accounting for the sequential design require approximately 300 children to compare the four embedded dynamic treatment regimens with 80 % power.
Case 2: Platform trial for diabetic macular oedema
Research question: Which anti-VEGF treatment strategies provide optimal visual outcomes with acceptable treatment burden in diabetic macular oedema?
Design: Bayesian adaptive platform trial with shared infrastructure evaluating multiple anti-VEGF agents and dosing regimens. Initial arms include standard monthly aflibercept, treat-and-extend aflibercept, faricimab, and a novel agent entering phase III testing. Response-adaptive randomisation allocates more patients to better-performing arms. Ineffective arms are dropped when posterior probability of inferiority exceeds 95 %, while new treatments can be added as they become available.
Estimand specification: A composite estimand defines success as gaining ≥ 10 ETDRS letters at 12 months without requiring more than 8 injections, capturing both efficacy and treatment burden within the primary endpoint.
Key considerations: Central reading centre grading of OCT and visual acuity ensures unbiased outcome assessment. The shared control arm improves efficiency while Bayesian updating enables continuous learning. Governance structures accommodate adding and dropping arms while maintaining trial integrity. Statistical analysis uses hierarchical Bayesian models borrowing information across similar treatment regimens.
Case 3: Hybrid decentralised trial for dry eye disease
Research question: Does a novel anti-inflammatory agent improve symptoms and signs in moderate-to-severe dry eye disease compared to standard artificial tears?
Design: Hybrid decentralised double-masked randomised controlled trial. Screening, baseline assessment, and visits at weeks 4 and 12 occur at clinical sites for slit-lamp examination, tear break-up time, and corneal staining. Daily symptom diaries (OSDI subscales) are collected via smartphone application. Weekly quality of life assessments use validated electronic patient-reported outcome measures transmitted directly to the study database.
Estimand specification: The primary estimand uses a treatment policy strategy for the symptom endpoint (mean change in OSDI at week 12) and a hypothetical strategy for the sign endpoint (corneal staining), estimating the effect that would have occurred had patients not used rescue artificial tears.
Key considerations: The hybrid approach enables more frequent symptom assessment than traditional site-based trials while maintaining rigorous clinical examination at key timepoints. Electronic data capture reduces transcription errors and enables real-time monitoring. Geographic reach expands beyond traditional academic centres while maintaining regulatory-quality data. Validated home visual acuity testing supplements site-based best-corrected visual acuity measurement.
Discussion
This comprehensive review demonstrates that clinical research methodology in eye and vision research has evolved substantially beyond the traditional parallel-group RCT paradigm. The diversity of approaches now available – from observational methods strengthened by target trial emulation to adaptive platform trials and SMART designs – enables researchers to match study design to specific research questions while maximising both efficiency and scientific rigour.
The unique challenges in eye research require careful consideration throughout the research process. The paired nature of eyes offers opportunities for efficient within-person comparisons but demands appropriate statistical methods to avoid inflated Type I errors. The difficulty of masking many ophthalmic interventions necessitates creative approaches to minimise bias, such as using masked outcome assessors, central reading centres, and standardised protocols. The long follow-up required for progressive diseases like glaucoma creates logistical and financial challenges that registry-based and pragmatic designs may help address.
The integration of decentralised trial elements represents an important evolution, expanding access to research participation while enabling more frequent outcome assessment. However, the specialised examinations fundamental to eye and vision research – including slit-lamp biomicroscopy, tonometry, OCT, and perimetry – remain site-based, making hybrid designs optimal for most ophthalmic research. The systematic review imperative before new trials ensures that research resources are directed toward genuine knowledge gaps, while updated reporting standards including CONSORT 2025 and the ACE statement ensure that methodological innovations are communicated with sufficient detail for replication and critical appraisal.
Looking forward, several priorities emerge for advancing clinical research in eye care. Enhanced statistical literacy through continued education for clinicians and researchers on appropriate design and analysis methods remains fundamental. Collaborative approaches through multi-centre studies and research networks achieve adequate sample sizes, particularly for rare conditions and for platform infrastructure. Patient-centred outcomes emphasising quality of life and functional outcomes meaningful to patients should guide endpoint selection. Methodological innovation in developing and validating new trial designs suited to ophthalmic conditions will continue advancing the field. Finally, transparency and reproducibility through adherence to reporting guidelines and data sharing practices maximise the value derived from research investments and participant contributions.
Conclusion
Rigorous clinical research design and analysis form the foundation of evidence-based eye care. This article has provided comprehensive guidance for selecting appropriate study designs, calculating sample sizes, implementing randomisation, choosing endpoints, and applying statistical methods specific to eye and vision research and optometry research. Critically, we have integrated traditional approaches with contemporary innovations including the estimands framework, platform trials, SMART designs, Bayesian adaptive methods, and decentralised trial elements.
The progression from observational studies identifying associations to randomised trials establishing causality represents a continuum of evidence generation. Each design serves important purposes, and the optimal choice depends on the research question, available resources, ethical considerations, and practical constraints. Non-randomised designs strengthened by target trial emulation and propensity score methods offer valuable insights when randomisation is not feasible, while modern experimental designs including adaptive and platform trials provide unprecedented efficiency for comparing multiple treatments.
Statistical considerations permeate every aspect of clinical research, from initial design through final analysis. Proper handling of paired eye data, appropriate methods for missing data, control of multiplicity through graphical approaches, and alignment of analysis with pre-specified estimands are essential for valid conclusions. The choice of endpoints should balance clinical relevance with measurement reliability, and analysis plans should be pre-specified to prevent data-driven decisions that inflate Type I errors.
As we advance toward precision medicine in eye care, the integration of genomic data, imaging biomarkers, and real-world evidence will require sophisticated analytical approaches while maintaining fundamental principles of good research practice. The ethical imperative to review existing evidence before initiating new trials, combined with updated reporting standards ensuring transparency, positions the field to generate robust evidence that improves patient outcomes. Building on the statistical foundations established in Parts 1 and 2 of this series, clinicians and researchers can contribute to evidence generation that advances our understanding of eye diseases while reducing research waste and accelerating translation to clinical practice.
The next article in this series will explore data visualisation techniques for effectively communicating research findings, while Part 5 will address the ethical dimensions of statistical analysis in eye research. Together, this series aims to enhance statistical literacy across the eye care community, ultimately supporting better research and improved patient care.
Conflict of Interest Declaration
The author declares that they have no affiliations with or involvement in any organisation or entity with any financial interest in the subject matter or materials discussed in this manuscript.
Funding Statement
This article did not receive a specific grant from public, commercial, or not-for-profit funding agencies.
Mayo-Wilson, E., Terwee, C. B., Chee-A-Tow, A., Baba, A., Gavin, F., Grimshaw, J. M., Kelly, L. E , Saeed, L., Thabane, L., Askie, L., Smith, M., Farid-Kapadia, M., Williamson, P. R., Szatmari, P., Tugwell, P., Golub, R. M., Monga, S., Vohra, S., Marlin, S., Ungar, W. J., Offringa, M. (2022). Guidelines for Reporting Outcomes in Trial Reports: The CONSORT-Outcomes 2022 Extension. JAMA, 328, 2252–2264.