Genetic counselling and gene analysis in patients with hereditary ocular diseases
Purpose: Non-syndromic hereditary ocular diseases can lead to severe functional limitations of vision at all ages. Therefore, molecular genetic research into the causes of hereditary ocular diseases is of paramount importance. Due to the increasingly optimised pathophysiologically adapted treatments and gene therapy options, a rapid determination of the genetic cause of hereditary ocular diseases is necessary, also to increase the chances of treatment at early stages of the disease. In this context, the authors retrospectively analysed the database of the “rare diseases” consultation hour of the PraxisKlinik Augenärzte Halle, which has been in existence since 2016, for patients with a suspected hereditary ophthalmological disease after molecular genetic diagnostics, counselling and therapy options.
Material and Methods: 367 index patients were treated during the period under consideration. In 221 (60.2 %) of them, the molecular genetic cause of the underlying disease could be found. In addition to a large group of patients with corneal dystrophies (304 patients, detection rate 61.2 %), we treated 23 index patients with hereditary retinal diseases (detection rate 69.6 %), 28 index patients with complex malformations (detection rate 50 %) and 15 index patients with optic atrophies (detection rate 33.4 %). In addition to the ophthalmological-clinical examination and diagnostics, genealogical analysis and molecular genetic testing were always performed.
Results: On the basis of 3 case studies with a molecularly genetically proven hereditary retinal disease (X-linked recessive inherited juvenile retinoschisis, hemizygous missense mutation in the RS1 gene; enhanced S-Cone syndrome, compound heterozygous mutation in the RS2E3 gene; oculocutaneous albinism, homozygous mutation in the TYP gene), the inheritance, the clinical picture, the course and the therapeutic and rehabilitative options were explained.
Conclusion: Ophthalmogenetic counselling and diagnostics in cases of suspected hereditary eye diseases are crucial for a prompt and a sufficient possible therapy and optical rehabilitation in affected families. Thorough history analysis and diagnostics can optimise the detection rate and provide rapid and sufficient patient care.
Introduction
Non-syndromic hereditary ocular diseases form one of the large, but clinically and genetically heterogeneous groups of rare diseases, which in many cases lead progressively to vision loss.
In recent years, genetic retinal changes in particular have become one of the major causes of blindness within the working population of the western world.1,2
A distinction is made between diseases of the cornea (corneal dystrophies), the optic nerve, retinal dystrophies, hereditary and juvenile glaucomas and complex ocular malformations (including aniridia, Axenfeld-Rieger anomaly) according to their anatomical location, and according to their course (stationary versus progressive).3,4
In addition to all forms of Mendelian inheritance (autosomal-dominant, autosomal-recessive, X-linked), there are also forms of mitochondrial or digenic inheritance.3
Due to phenotypic similarities within the individual groups, a clinical diagnosis is not always possible in many cases. For this reason, additional molecular genetic testing is an essential component for confirming the diagnosis and in some cases also for planning therapy.3,4,5
The development in recent years of Next Generation Sequencing (NGS) technologies with its high sequencing capacities enables simultaneous and cost-effective analysis of a large number of candidate genes through parallel sequencing of gene panels and thus leads to an increase in the identification rate of the underlying genetic defect in hereditary ocular diseases.5,6,7,8 In certain cases (with a clearly defined clinical diagnosis and recurrent monogenic cause of the ophthalmic disease in question), targeted conventional Sanger sequencing is initially preferable to a broad genetic investigation using multigene panel analyses or whole exome or whole genome sequencing.9,10
This always presupposes a reliable clinical diagnosis and an exact analysis of the inheritance, which can be decisively improved by joint care in centres specialising in rare diseases and an intensive interdisciplinary exchange with human geneticists with specialist experience.
Sufficient clinical and molecular genetic diagnosis is indispensable for the following reasons: it is a prerequisite for genetic counselling, among other things for family planning; it improves disease management and helps with counselling regarding the course of the disease; in many cases it can save further (also time- and cost-intensive) examinations and it is a mandatory prerequisite in the case of therapy planning, also with regard to gene therapy.10,11
In our consultation hour for rare diseases, which has existed since 2016 and in which we carry out diagnostics, counselling and control examinations, we treat patients with hereditary ocular diseases. Within the framework of a retrospective evaluation, we analysed 367 cases from our database with a suspected hereditary ophthalmological disease and a molecular genetic diagnosis. In this paper, we would like to present an overview of the spectrum of diseases treated, the detection rate of the underlying genetic alteration and the course of the disease of selected cases.
Methods
Patients and clinical diagnosis
Within the framework of a „rare diseases“ consultation, which we carry out as a member of the DRN-eye network (German Reference Network for Rare Eye Diseases), we treat between 50 and 80 index patients and their relatives for diagnostics, counselling, therapy and follow-up at the PraxisKlinik am Markt, Halle every year. Our focus is on counselling and treatment of diseases of the cornea (especially corneal dystrophies) and the anterior segment of the eye, but also of diseases of the optic nerve and the retina (hereditary retinal dystrophies, IRD). All patients‘ data were continuously documented in our database independent of the clinical diagnosis. A molecular genetic examination was always performed if the patient agreed to this examination in writing after detailed counselling. In addition, written consent has been documented for each patient in accordance with the principles of the Declaration of Helsinki.
The ophthalmological clinical examination was performed as follows, depending on the underlying disease:
- For all patients: Autorefractometer, best-corrected distance visual acuity, applanation tonometry according to Goldmann, slit-lamp biomicroscopy, fundus examination in mydriasis.
Additional and individualised:
- In patients with diseases of the anterior segment of the eye: OCT of the anterior segment, corneal topography, corneal sensitivity, slit lamp photography.
- In patients with diseases of the retina and optic nerve: Colour vision testing (panel D15), computerised perimetry, twilight vision, OCT of the retina and optic nerve, fundus photography and fundus autofluorescence, in selected cases sensory examinations (ERG, multifocal ERG, VEP).
Genetic analysis
A family tree was drawn up for each patient and their family. In accordance with the requirements of the Gene Diagnostics Act, all those seeking advice for predictive diagnostics and all children were presented at a centre for human genetics. For molecular genetic diagnostics, EDTA blood tests were taken for DNA extraction, or, in the case of children, a cheek swab was taken during counselling by a human geneticist. Depending on the clinical diagnosis made, either Sanger sequencing of a single gene (Fuchs endothelial dystrophy, individual forms of corneal dystrophies) or panel diagnostics were performed. In individual cases, in the absence of a molecular genetic diagnosis, further candidate genes were analysed.
All findings were discussed in detail with the patients and families and in all cases, counselling with a human geneticist was offered.
Results
In 221 of 367 index patients, the molecular genetic cause of the underlying disease could be determined (Table 1), which corresponds to a detection rate of 60.2 %. The largest group consisted of patients with corneal diseases (corneal dystrophies; 304 analysed patients, detection rate 61.2 %), followed by complex ophthalmological malformations (28 analysed patients, detection rate 50 %), retinal diseases (retinal dystrophies, 23 analysed patients, detection rate 69.6 %) and diseases of the optic nerve (15 analysed patients, detection rate 33.4 %).
The group of retinal dystrophies will be discussed in more detail in the following section.
Compared to the other groups and in particular to the optic atrophy group, the detection rate of the underlying molecular genetic cause was highest here, but it was the most heterogeneous group based on molecular genetic cause with underlying mutations in the following genes: BEST1, RP2, ABCA4, PRPH2, RS1, CDHR1, USH2A, PAX6, TOPORS, BBS1, CHM, NR2E3 and CRX.
We would like to report on 3 cases in more detail below:
Case report 1
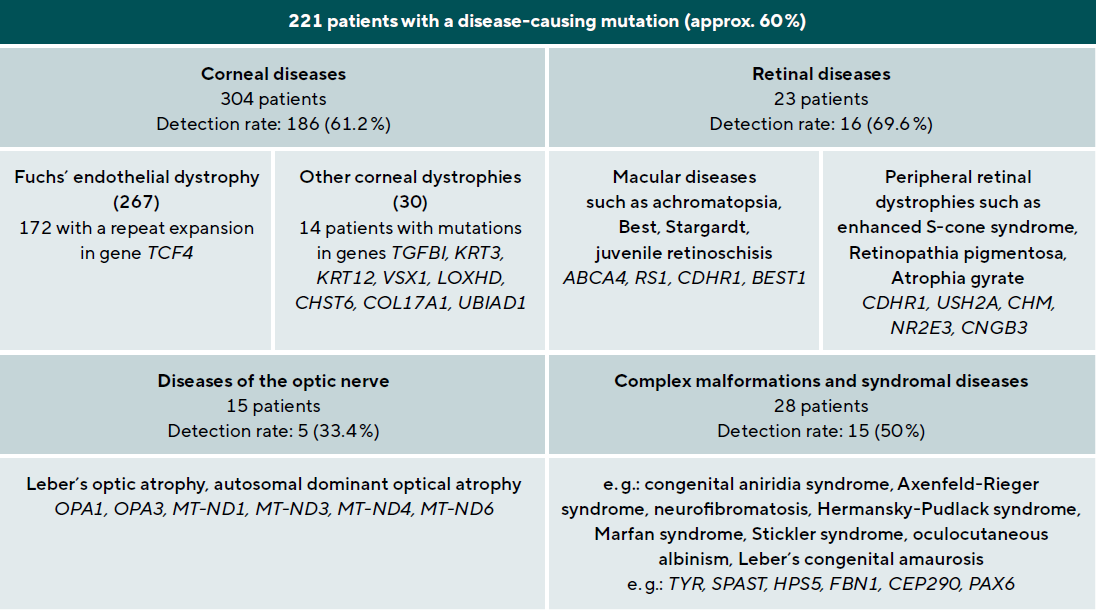
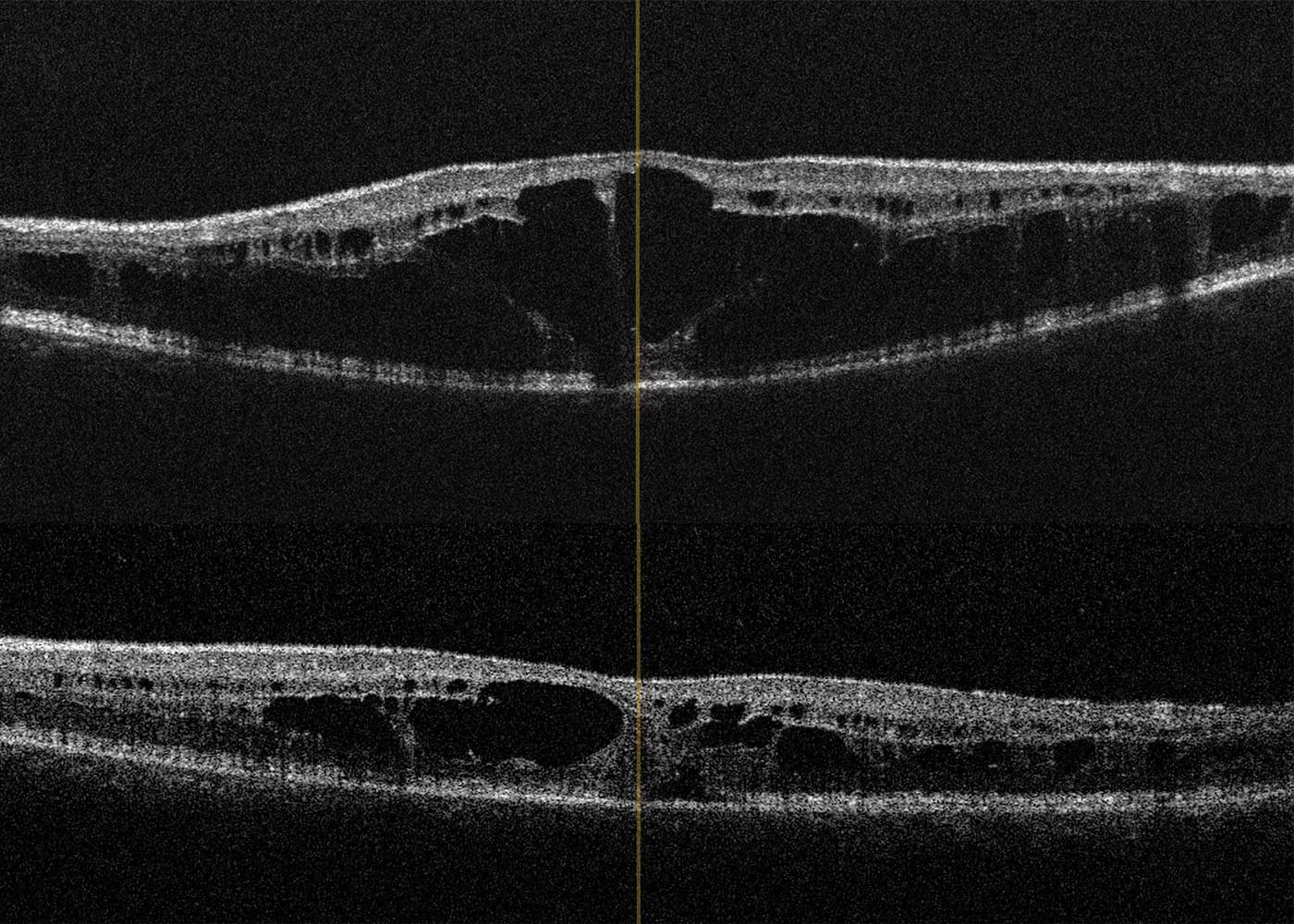
A 78-year-old patient came to the clinic for the first time and reported a visual change that had existed since childhood, and which had not been further clarified so far. The primary reason for coming was a visual defect of his 8-year-old grandson, which had now also been diagnosed. The visual acuity of the index patient was 0.3 on the right and 0.2 on the left eye. Funduscopic examination in mydriasis revealed bilateral diffuse pigment epithelial dystrophy and atrophy of the central and peripheral retina with no preference for one or the other area and no abnormal optic disc findings. Optical coherence tomography of the central retina showed foveal atrophy on both sides with indistinct retinal layers and single cystoid structures as well as unspecific atrophy mainly of the nerve fibre layer, the ganglion cell layer and the photoreceptors of the foveola. In addition, an atypical epiretinal gliosis and an unusual separation of retinal tissue in the area of the internal limiting membrane and the nerve fibre layer were seen (Figure 1).
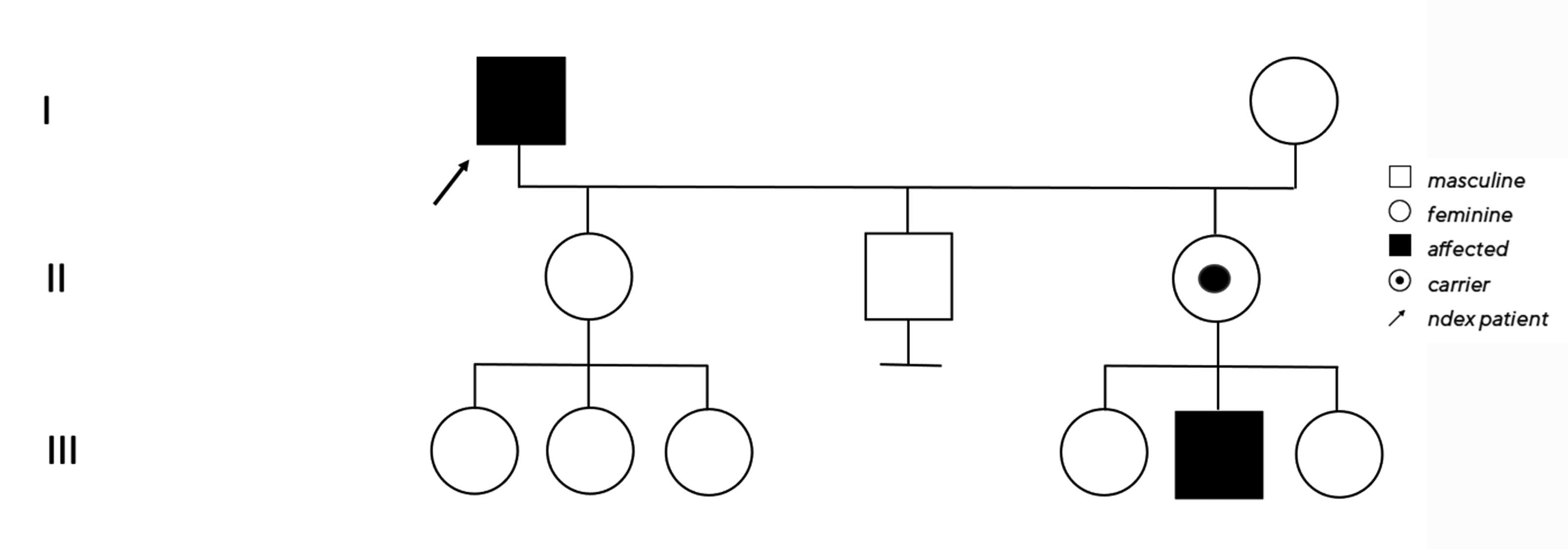
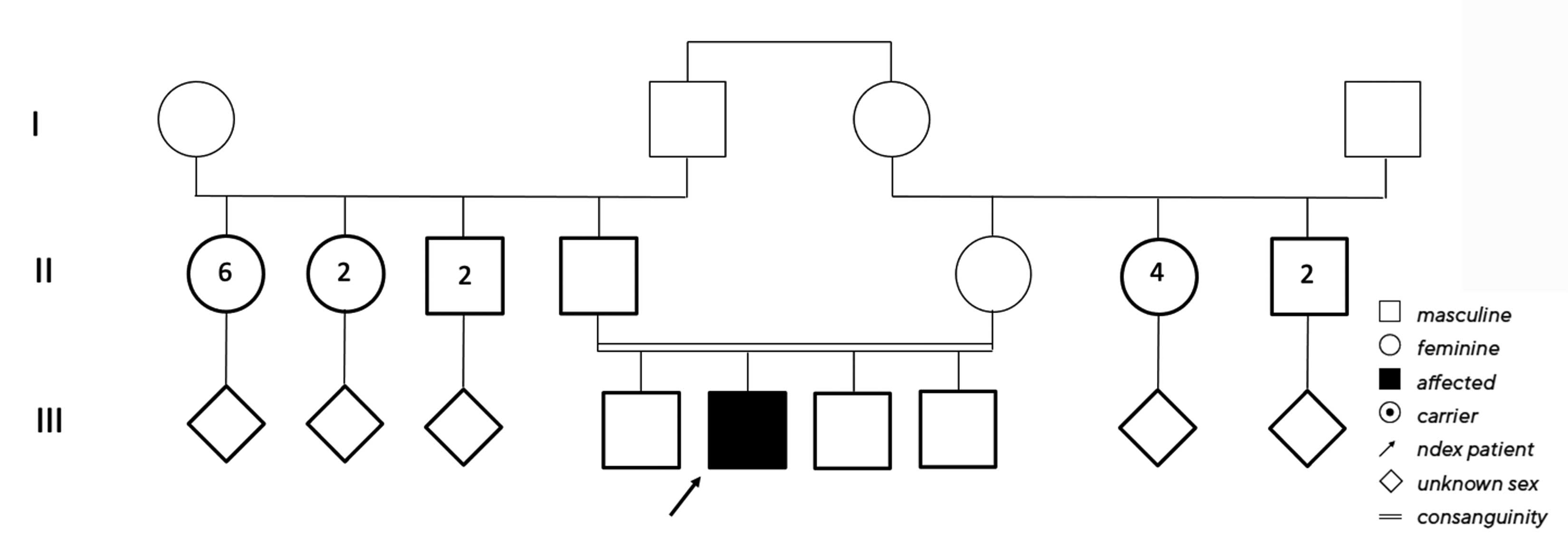
No clear diagnosis of retinal dystrophy could be assigned to these unspecific changes after the initial examination. In a further consultation, the grandson was clinically examined with a visual acuity of 0.2 on the right and 0.3 on the left and a photographically documented star pattern of the macula on funduscopy (Figure 2a). We could not find a pigment epithelial dystrophy as seen in the grandfather and the optic disc findings were unremarkable. On OCT examination of the grandson‘s foveola, we found a typical splitting of the intraretinal nerve fibre layers in the sense of a foveal retinoschisis without signs of a peripheral retinoschisis (Figure 2b). The mother of the child (daughter of the index patient) was ophthalmologically healthy with a visual acuity of right/left 1.0 and unremarkable OCT findings. Other family members were not affected (genealogy, Figure 3). The family was examined at the human genetics consultation, where a panel diagnosis (Sanger sequencing, panel macular dystrophy) was initiated in the index patient and the affected grandchild, based on the course in the grandfather, the clinical findings in the child and the inheritance derived from the genealogical analysis (X-linked recessive) with the suspected diagnosis of X-linked juvenile retinoschisis.
The hemizygous missense mutation c.487T>G with an amino acid substitution p.Trp163Gly in the RS1 gene on the X chromosome was found in both affected family members. Subsequently, the unaffected daughter was examined molecular-genetically as a presumed carrier and we also found the above-described heterozygous change on one of the two X chromosomes.12
In X-linked juvenile retinoschisis (incidence 1 : 500 – 1 : 25000), there is a symmetrical splitting of the retina, which manifests in the first decade of life by a progressive loss of visual acuity. The consequences can be retinal detachments or vitreous haemorrhages. Due to the X-linked inheritance, usually only male family members are
affected.13,14
The RS1 gene is located on the short arm of the X chromosome and encodes the retina-specific cell-adhesion protein retinoschisin 1. It is secreted into the intercellular space by the photoreceptor and bipolar cells of the retina. Defects or an absence of the protein leads to a lack of maintenance of cell-cell interaction and cell adhesion of the inner retina and thus to the development of intraretinal microcysts with increasing splitting of the inner retinal layers. The consequence is a disorganisation of the bipolar cells and a disturbance of the synaptic transmission.15,16,17
A curative therapy is not yet known; important is the provision of optical aids, optimal schooling and suitable career planning. The index patient did not want optical or rehabilitative care.
Case report 2
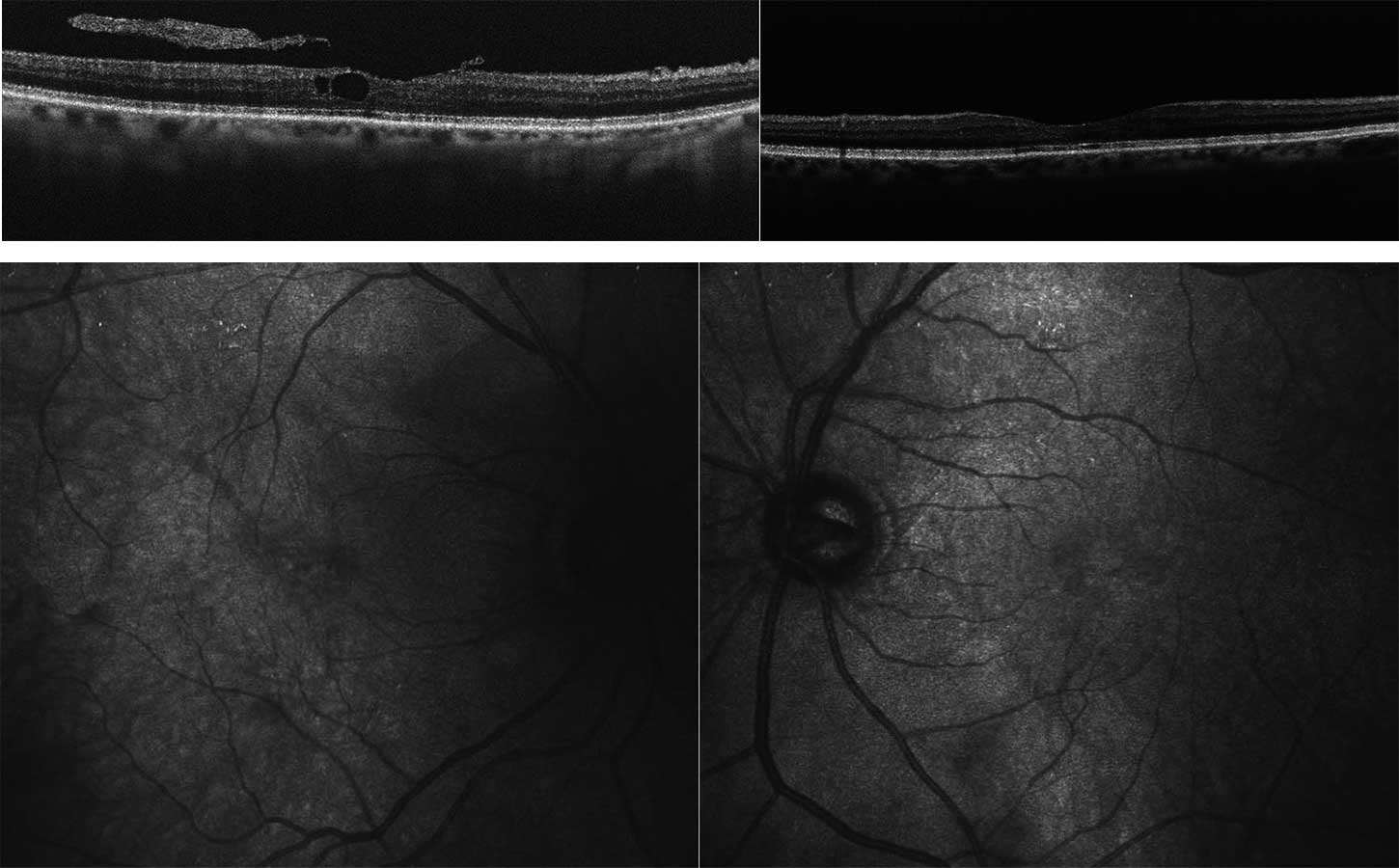
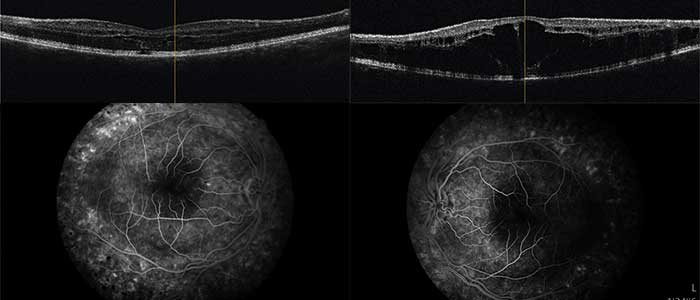
A 14-year-old boy and his parents came to the clinic for the first time for a co-assessment of increasing visual deterioration on both sides. He had been examined regularly by an ophthalmologist, who detected non-specific progressive retinal changes which had not been further clarified. During the first appointment, the visual acuity was 0.6 on the right and 0.1 on the left eye. Funduscopic examination revealed pigment epithelial clumps of the peripheral retina in the form of bone trabeculae on both sides and fluorescence angiography revealed a discrete but clinically significant diffuse macular oedema. In contrast to the findings described in case 1, OCT of the macula showed large, bubbled cystoid changes on both sides with angiographically proven intraretinal fluid as oedema (Figure 4). The colour vision test panel D15 was unremarkable and there were no changes in the outer visual field borders on either side. No other affected family members were found in the family history. Here, too, the patient was brought to the human genetics clinic and a panel diagnosis (Sanger sequencing) was initiated for suspected rod-cone dystrophy in the index patient.
Two compound heterozygous pathogenic variants were found in the RS2E3 gene: The missense mutation c.311G>A, which leads to an amino acid exchange p.Arg104Gln on one allele and the nonsense mutation c.309C>A, which leads to a stop mutation p.Cys103* on the second allele. Both mutations have already been described as causing the disease both in the autosomal dominant inheritance, here for retinopathia pigmentosa, and in the autosomal recessive mode of inheritance, here for enhanced S-cone syndrome.
Clinically, these mutations lead to a reduction and degeneration of the red and green cones with a simultaneous excess of S cones (blue). Characteristics are red-green weakness, early visual deterioration with night blindness, visual field impairment and hypersensitivity to blue light. This form of retinal dystrophy can progress with age. Morphologically, peripheral retinochoroidal dystrophy with bone corpuscles and in some cases the cystoid maculopathy described here are found.18,19
The RS2E3 gene is located on the long arm of chromosome 15 (15q23) and belongs to a large group of nuclear receptor transcription factors that are responsible for the regulation of signalling pathways, especially in cone differentiation during embryogenesis. Mutations lead to an under-regulation of cone differentiation. The result is an excess of S cones (blue) with a clear under-representation of red and green cones, which explains the hypersensitivity to blue light with simultaneous red-green weakness. Due to its clinical picture, the enhanced S-cone syndrome is not infrequently misdiagnosed as retinopathia pigmentosa or as X-linked juvenile retinoschisis.20,21
Due to the fluid-filled intraretinal cysts found angiographically, a therapy with a carbonic anhydrase inhibitor (initially systemic, after 4 weeks as local therapy) was started.22 This resulted in a visual acuity gain to 0.4 in the left eye with visual stabilisation in the right eye due to a significant reduction in intraretinal fluid, especially on the left (Figure 5). At the same time, electronic equipment for digital magnification were arranged for the school in addition to special workplace lighting.
Case report 3
A 5-year-old Syrian boy came to the clinic with his parents with an external diagnosis of ocular pendulum nystagmus and bilateral amblyopia with high hyperopia (refraction in cycloplegia bilaterally: +8.0 D) with the recommendation to undergo occlusion therapy.
The visual acuity during the first consult was 0.1 in the Lea test at 3 m uncorrected with previously unadjusted glasses on the right and left eyes. After adjustment using fully corrected glasses, the visual acuity could be increased to 0.3 (Lea test 3 m) on both sides after 6 weeks, an improvement of visual acuity at 5 m was not possible.
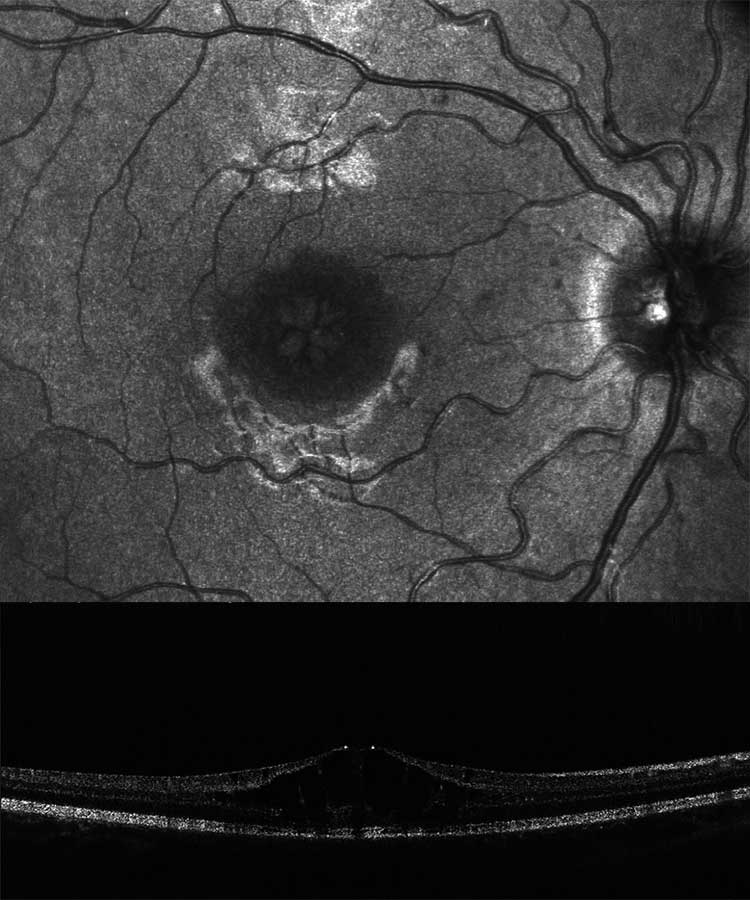
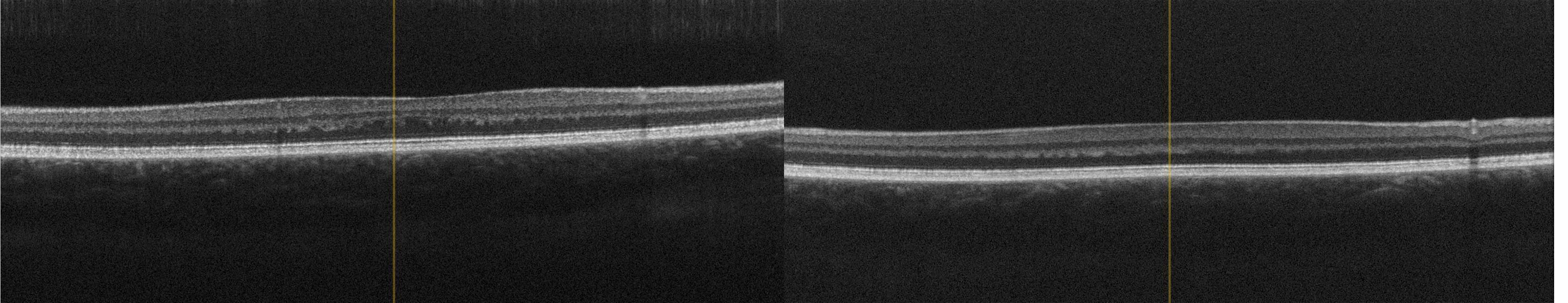
A genealogy survey revealed consanguinity of the parents (cousins, Figure 6). Foveolar aplasia was seen on both sides in the subsequent OCT examination of the macula (Figure 7). Both optic discs were regular in size and nerve fibre thickness. After a first consult in the human genetics clinic, a panel diagnosis (Sanger sequencing) was initiated in the index patient since macular dystrophy was suspected. A homozygous pathogenic variant was found in the TYR gene: The missense mutation c.1264C>T causes an amino acid exchange p.Arg422Trp on both alleles of the TYR gene. This mutation has already been found to cause an autosomal-recessive disease, causing oculocutaneous albinism type 1 with foveolar hypo-/aplasia.23,24
The TYR gene is located on the long arm of chromosome 11 (11q14-21), consists of 5 exons and codes for the protein tyrosinase. Mutations in the TYR gene cause a form of oculocutaneous albinism type 1 and lead to either a complete (tyrosinase-negative form) or a partial loss of function (tyrosinase-positive form). The consequence is always a disturbance of melanin synthesis on the cell wall of the melanocytes with a complete or partial absence of melanin.23,24,25,26 In addition to a possible hypopigmentation of hair, skin and iris, the visual system of affected individuals shows foveolar hypoplasia with consecutive sensory nystagmus due to a developmentally reduced, topographically redistributed number of photoreceptors at the posterior pole of the retina and, as a consequence, an insufficiently mature formation of the macula. Optic dysplasia is found in many cases, particularly in oculocutaneous albinism type 1. In addition, there is an atypical chiasmal crossing of the optic nerves.26,27
Due to the molecular-genetically confirmed diagnosis, after optimal spectacle fitting, occlusion treatment was discarded based on the equal visual acuity on both eyes, which was explained by the underlying disease, and schooling in a specialised institution with special educational support was arranged. In addition, and considering the recurrence risk of 25 %, the siblings were also examined. Clinically and ophthalmologically, there was no evidence of oculocutaneous albinism in the other children.
Recommendations on how to proceed in cases of suspected hereditary ophthalmic disease
If there is a discrepancy between function (visual acuity/field of vision) and clinical examination results and if the patient‘s overall morphology is conspicuous, a detailed family history including a genealogical survey should be undertaken. In addition, further examinations (including OCT of the anterior and posterior segment of the eye, colour vision tests, fundus autofluorescence and fluorescence angiography, electrophysiological examinations) should be performed depending on the affected structure. This makes it possible to narrow down the clinical picture. If a genetic cause is suspected, referral to a specialised ophthalmological centre (listed in the German Network for Rare Eye Diseases DRN-Eye or in the German National Action Alliance for Rare Eye Diseases NAMSE) is advisable. It is important to have as exact a suspected diagnosis as possible when making an appointment at a human genetics facility in order to be able to precisely narrow down the gene panel to be examined.
After diagnosis, apart from referral to clinical centres, for example to a gene therapy centre, visual rehabilitation with optical and technical aids, learning compensation techniques (e.g., reading training), social counselling (including educational, vocational and financial security) and psychosocial counselling are necessary. Vocational support centres, low-vision centres and rehabilitative inpatient facilities are available for this purpose.
Discussion
Rare diseases, which are often hereditary, represent a considerable challenge for a reliable diagnosis and subsequent therapy planning for the treating ophthalmologist because of their great genetic heterogeneity, the overlapping clinical signs and the variability of inheritance patterns.3,5,8,11 Using three case studies of patients exhibiting hereditary retinal diseases, we have presented different clinical findings, different modes of inheritance and the resulting possible therapeutic and rehabilitative options and the possible consequences for the families.
In case report 1, a definite clinical diagnosis was not possible due to the advanced findings of the index patient alone; the genealogy here showed the distinct X-linked recessive inheritance and, together with the clinical findings of the grandson, considerably limited the molecular genetic options. It also shows the considerable variable expressivity of the variant within a family. Previous case reports have also failed to document a clear correlation between genotype and phenotype.13,14 Many studies of X-linked juvenile retinoschisis have documented the increasing retinal thinning seen here with age, with fusion of the retinal layers due to reduction of the retinal cleft and subsequent increasing irregular foveal contour.12,14,16 Surprisingly, the visual acuity of the index patient was comparable to that of the grandson despite these significant changes over the course of his life. In the meantime, the first studies have been conducted on this clinical picture using gene therapy on mice,28,29 so we referred the family under treatment to a centre specialising in gene therapy. For the grandson, in addition to visual rehabilitation, we also made a social-pedagogical referral.
In contrast to this case report, in case report 2 there were no ground-breaking indications due to a conspicuous inheritance. In this case, the molecular genetic panel diagnostics was helpful due to the cone-rod dystrophy, which could be diagnosed with clinical certainty.20,21 Whereas in case report 1 the cleft formation of the foveola was caused by a disorder of cell-cell interaction and cell adhesion of the inner retina, in this case report fluorescence angiography demonstrated clinical macular oedema (Figure 4b). After referral to a centre experienced in gene therapy for hereditary retinal dystrophies, a therapy with carbonic anhydrase inhibitors was started, which at the present time has resulted in an improvement in the findings.22
In case report 3, no other affected family members were indicated in the first clinical history. However, the consanguineous nature of the parents was indicative of a genetically caused disease (intermarriage, see Figure 6), which significantly increased the risk of an autosomal recessively inherited disease (in this case, the foveolar hypoplasia found in the OCT). For this reason, a molecular genetic examination (panel diagnostics, macular dystrophy) was performed.
The mutation in the TYR gene found here, resulting in oculocutaneous albinism type 1, is responsible for 40 % of all forms of albinism and is thus the second most common cause of the condition.24,26 The visual acuity in this form is between 0.1 and 0.2 in most patients, whereby the cause of the relatively poor visual acuity compared to other forms of albinism is to be found in the frequency of optic dysplasia with over 50 % in this form of albinism.27 Despite the lack of curative therapy options, function and integration could be optimised through adjusted spectacle fitting with a visual acuity increase from 0.1 (Lea test, 3 m) on both sides to 0.32 (Lea test, 3 m) and through socio-paediatric care with special education. Intensive human genetic counselling in case of a further wish to have children (risk of recurrence 25 %) and the control of siblings is indispensable and was also arranged.
We can show with our examples that the diagnosis of hereditary ocular diseases has been considerably simplified by new technological test possibilities. An exhaustive family history is indispensable in case of suspicion, as are an exact knowledge of underlying pathological findings for correct classification and a trusting and collegial cooperation with a competent human genetics centre. In many cases, this can avoid costly additional examinations and an early initiation of rehabilitative but also therapeutic procedures can be provided.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the methods and devices mentioned in the article.
Pictures: Copyright by Claudia Grünauer-Kloevekorn
