Ocular manifestations of neurodegenerative demyelinating diseases
Purpose: Central nervous system (CNS) demyelinating diseases are one type of neurodegenerative disease that can progressively impact the visual system. Although their name focuses on the reversible myelin damage, these conditions also demonstrate slowly progressive permanent neuronal damage, earning them the categorization as neurodegenerative diseases.
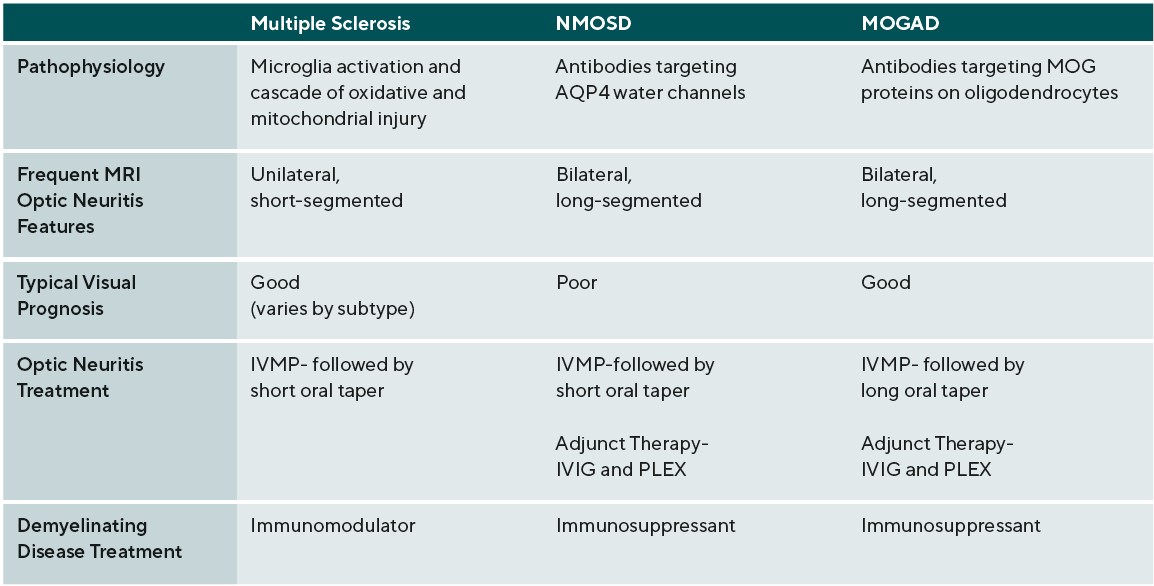
Material and Methods: Neurodegenerative demyelinating diseases of the central nervous system (CNS) include multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD), and myelin oligodendrocyte glycoprotein antibody disease (MOGAD). Although all involve inflammation and axonal loss, the mechanisms underlying MS, NMOSD and MOGAD differ; subsequently, their clinical presentations, diagnostic features, and pharmacologic treatments also vary.
Results: Neurodegenerative demyelinating diseases can affect both the afferent and efferent visual system, prompting patients to seek care from eye care providers often due to vision loss or diplopia. Specifics of the clinical presentation can help not only in the initial suspicion of a neurodegenerative demyelinating disease, but also in distinguishing among them. Since the optic nerve is often affected, optical coherence tomography (OCT) can be a helpful biomarker to assess disease activity and progression.
Conclusions: Eye care providers need to be aware of the similarities and differences in the ocular manifestations of neurodegenerative demyelinating diseases in order to aid in early diagnosis and appropriate management, which can in turn affect prognosis. Eye care providers are a vital part of the care team and need to remain updated on the latest advances regarding CNS demyelinating diseases.
Introduction
Neurodegeneration is a general term referring to progressive damage and/or death of neurons, and can be a feature of many different disease processes. The term neurodegenerative disease is often correlated with age-related conditions, such as Alzheimer disease and parkinsonian syndromes. However, neurodegenerative processes are not unique to the elderly population. That is especially true for the neurodegenerative demyelinating diseases of the central nervous system (CNS), such as multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD), and myelin oligodendrocyte glycoprotein antibody disease (MOGAD). Demyelination, a defining feature of these diseases, occurs due to an immune process resulting in acute inflammation; but myelin is not the only structure affected. These conditions also share a slowly progressive component of neuronal damage. Whereas myelin can regenerate, neurons of the CNS are unable to do so, resulting in neurodegeneration. While it is not completely understood whether neurodegeneration in the CNS is a primary process or is secondary to inflammation, it results in many of the ophthalmic manifestations and consequences in these demyelinating diseases. Since the optic nerve, and the axons of retinal ganglion cells which comprise it, are part of the CNS, the afferent visual system is commonly affected in these demyelinating diseases. Optical coherence tomography (OCT) is a unique tool that eye care providers can use to appreciate and quantify neuronal damage of the afferent visual system. The efferent visual system can also be impacted when the nerves are damaged before leaving the CNS and becoming part of the peripheral nervous system (PNS). Eye care providers need to be aware of the similarities and differences in the ocular manifestations of neurodegenerative demyelinating diseases in order to aid in early diagnosis and appropriate management, which can in turn affect prognosis. This article reviews the pathophysiology, clinical manifestations, diagnostic criteria, and treatment options of these CNS demyelinating diseases.
Pathophysiology
The chronic inflammatory nature of all neurodegenerative demyelinating diseases results in axonal loss; however, the mechanisms underlying MS, NMOSD and MOGAD differ; subsequently, their clinical presentations, diagnostic features, and pharmacologic treatments also vary. MS is characterized by focal inflammation, demyelination, reactive gliosis, and secondary axonal degeneration.1,2 Although the exact cause initiating the inflammatory process is unknown, complex genetic traits and environmental factors influence the susceptibility of disease development.2,3 The mechanism ultimately responsible for demyelination and degeneration is inflammation leading to microglia activation, which creates a cascade of oxidative injury and mitochondrial damage, resulting in hypoxia. 4,5 The inflammation specifically surrounds post-capillary venules and veins. Therefore, active lesions composed mainly of microglia and CD8+ T cells, along with a smaller amount of CD4+ T cells, B cells and plasma cells, form in the perivenous region. These lesions become confluent demyelinated plaques which lead to loss of the oligodendrocytes responsible for production of myelin in the CNS. 1,5 At later stages of the disease, gliotic scars form and there is widespread demyelination and degenerative changes in both white and gray matter.3
It is of note that the clinical course of MS is heterogeneous, and thus, while these patients share the above pathologic features there are also some physiologic differences. Specifically, MS can be categorized into three clinical courses: relapsing-remitting MS (RRMS) is the most common and is defined by clinical relapses followed by functional recovery, secondary-progressive MS (SPMS) is a progressive stage that RRMS patients may enter, and primary-progressive MS (PPMS) follows a progressive course at onset.1 NMOSD is associated with CNS demyelination distinct from MS and is an astrocytopathy associated with the presence of aquaporin-4 immunoglobulin G antibodies (AQP4-IgG). AQP4 is the most abundant water channel in the brain, spinal cord, and optic nerve.6
It is expressed on astrocytic foot processes that surround CNS blood vessels and the subarachnoid space, and is involved in transcellular water transport.7 The mechanism for NMOSD astrocyopathy starts with AQP4 peptides circulating outside the CNS being recognized by naïve T cells, and becoming polarized to helper T-cell subtype 17 (Th17) phenotype which help B cells that have been activated by AQP4 antigens. Interleukin-6 (IL-6) promotes differentiation of these AQP4-dependent activated B cells into plasmablasts, which secrete AQP4-IgG.8 The penetration of AQP4-IgG through the blood-brain barrier (BBB) to the CNS may be a result of intrathecal production of AQP4-IgG initiating focal BBB breakdown precipitating an influx of serum AQP4-IgG or systemic inflammation disrupting the BBB.9 It is also thought that activated AQP4-specific T cells play a role in allowing the entry of AQP4-IgG into the CNS, as Th17 cells mediate the disruption of the BBB. 7,9 Once in the CNS, the binding of AQP4-IgG to AQP4 on the astrocytic foot processes then leads to CNS injury with the activation of the complement cascade, granulocyte and macrophage infiltration, secondary oligodendrocyte damage, and neuronal death.5,7,9 It is of note that there are patients with the clinical features of NMOSD who are seronegative for AQP4-IgG. A subgroup of these patients are seropositive for myelin oligodendrocyte glycoprotein immunoglobulin G antibodies (MOG-IgG); and are now categorized as MOGAD.10,11,12
MOGAD, as its name suggests, is a condition in which antibodies target myelin oligodendrocyte glycoproteins on the surfaces of oligodendrocytes. It includes a broad clinical phenotype, including AQP4-IgG negative NMOSD, acute disseminated encephalomyelitis (ADEM), optic neuritis (ON), myelitis, and encephalitis.13 MOG is primarily expressed within the brain, spinal cord, and optic nerves. MOG is produced by oligodendrocytes and plays a role in the formation, maintenance, and disintegration of myelin sheaths. Furthermore, MOG may play a role in the adhesion between neighboring myelinated fibers.14 MOG is expressed on the outermost location of myelin sheaths, which makes it more easily accessible to antibodies and T-cells.14 The penetration of MOG-IgG through the BBB may be a result of infection, which is frequently associated with ADEM.13 The resultant autoimmunity leads to perivenous and confluent white matter demyelination.14 Since MS, NMOSD, and MOGAD all lead to demyelination and neurodegeneration of the CNS, there may be some overlap in their presentations. Nonetheless, each is a distinct spectrum of conditions and there are key ocular symptoms and features that may help distinguish them. Given the distinct pathophysiology of each condition, targeted therapeutics are indicated as treatment for one condition may be ineffective for, or even worsen, another.
Clinical Presentation
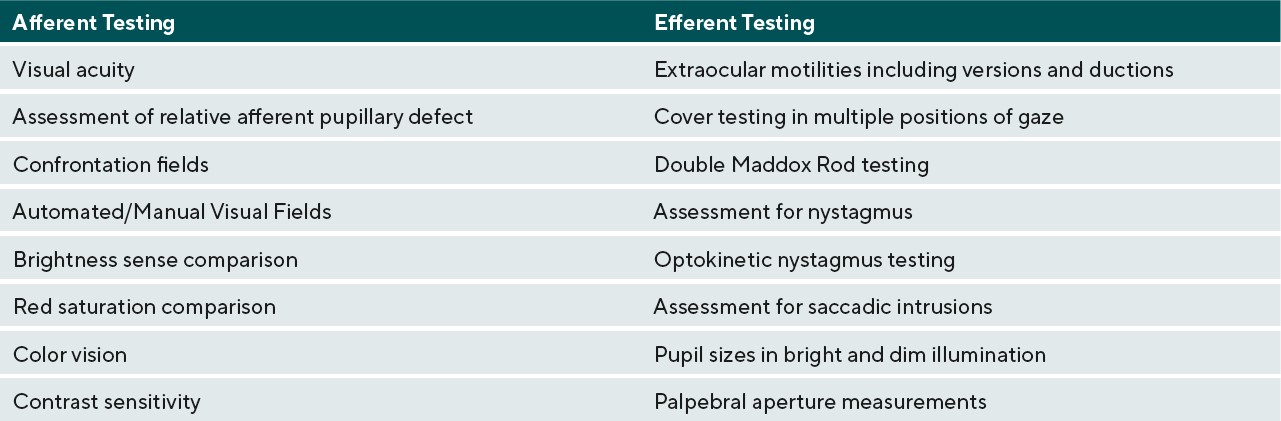
Effects of neurodegenerative demyelinating disease on the visual system can be debilitating and patients will seek care from their eye care providers to help improve their quality of life. In demyelinating conditions, it is the anatomical location of the acute inflammation and more chronic axonal loss that determines the specific subsequent ocular manifestations affecting the afferent and/or efferent visual systems.15,16 Table 1 lists clinical tests to assess afferent and efferent visual function. Afferent issues such as decreased acuity and contrast sensitivity can be addressed with the use of magnification and contrast-enhancing tints in glasses. Efferent system issues causing diplopia can be addressed with the use of Fresnel prisms in an acute situation and ground-in prism in more chronic diplopic situations.
Efferent visual System
Accurate conjugate eye movements and gaze-holding require intact complex neuronal pathways. Demyelinating disease affecting the brainstem, cerebellum and cerebrum may manifest as ocular motor deficits such as cranial nerve palsies, internuclear ophthalmoplegia (INO), bilateral internuclear ophthalmoplegia (BINO), wall-eyed bilateral internuclear ophthalmoplegia (WEBINO), skew deviation, and nystagmus. Damage to the abducens nerve and/or nucleus results in an ipsilateral abduction deficit or ipsilateral horizontal gaze palsy, respectively.15,17 Involvement of the oculomotor nerve is typically partial and may occur with or without pupil involvement and ptosis. Unilateral ptosis is most often associated with axonal lesions whereas bilateral ptosis may signify nuclear lesions.16,17 Trochlear nerve involvement is rare because the course of the nerve is short within the CNS and, therefore, has limited myelin produced by oligodendrocytes.16 Once cranial nerves leave the brainstem and are part of the PNS they are not affected by CNS demyelinating conditions.
INO resulting from a demyelinating plaque found in the medial longitudinal fasciculus (MLF), is the most common hallmark efferent feature of MS . The MLF is a brainstem interneuron pathway that regulates horizontal gaze by connecting the abducens nucleus to the contralateral oculomotor nucleus .16 Damage to the MLF in the midbrain or pons results in an INO, or limited adduction of the ipsilateral eye and an abducting nystagmus of the contralateral eye. This can often be a subtle finding, which may be more easily appreciated by looking for asymmetric optokinetic nystagmus. Involvement of the midbrain MLF can also affect convergence, while convergence is spared in lesions involving the pontine MLF. Damage to the MLF on both sides of the brainstem results in a BINO. WEBINO can occur with damage to the bilateral medial rectus subnuclei in the midbrain, resulting in a large exotropia, loss of convergence, and upbeat nystagmus.15 Similarly, one-and-a-half syndrome (an ipsilateral gaze palsy and contralateral INO) may result from a lesion of the pontine tegmentum damaging the pontine paramedian reticular formation or the abducens nucleus and ipsilateral MLF.18
Another clinical presentation often seen in demyelinating diseases and occurring concurrently with INO is skew deviation. Features of skew deviation occur because of damage to the graviceptive pathway and can include vertical ocular misalignment, head tilt and ocular torsion. This pathway connects vestibular nuclei to the nuclei of cranial nerves that will innervate the extraocular muscles. Therefore, a classic pattern is often seen with the higher eye intorted, the lower eye extorted, and head tilt away from the higher eye. Because this pathway crosses in the low pons, after leaving the vestibular nuclei, the higher eye will either be ipsilateral to a lesion above the crossing or contralateral to a lesion below the crossing.17 Upbeat nystagmus is associated with BINOs.15 Downbeat nystagmus is typically seen in lesions involving the cervicomedullary junction or cerebellum.15 Acquired pendular nystagmus is seen with lesions of the pathways that connect the brainstem networks and the cerebellum.17 Other efferent findings include abnormal saccades or saccadic intrusions. Small degree square-wave jerks may arise form a lesion of the pause cell neurons on the pontine raphe. Additionally, hypermetic or hypometric saccades may present with a lesion of the dorsal vermis, posterior fastigial nuclei or a lesion within the cerebellar peduncles.17
Afferent visual System
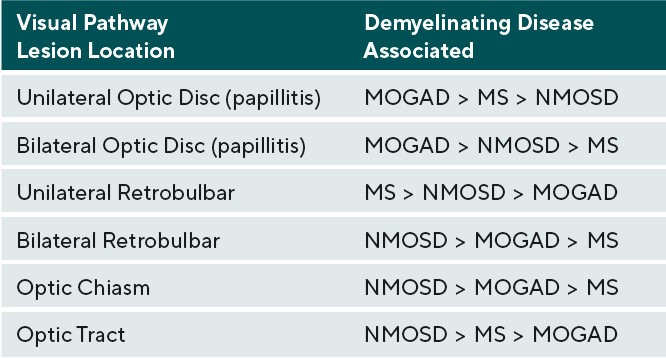
Afferent visual system damage occurs more frequently than efferent damage, with ON being the most common ocular presentation. Symptoms of ON include pain on eye movement, sudden loss of visual acuity and/or visual field, and dyschromatopsia.19 Pain on eye movements can present prior to any subjective visual changes. Once vision loss does occur, a relative afferent pupillary defect can be present in unilateral or asymmetric cases. These symptoms often improve when the active inflammation has ceased; however, there may be residual visual deficits secondary to past damage.20 Visual acuity is commonly affected in ON during active inflammation. The degree of post-inflammatory visual recovery depends on the specific demyelinating condition. In ON patients, recovery was worse in NMOSD and the greatest in MOGAD followed by MS.20 AQP4-IgG positive NMOSD patients were most likely to continue to have poor vision after inflammation waned.20 Unilateral ON is most common in patients with MS compared to NMOSD and MOGAD. The optic disc can appear swollen(papillitis) or normal in cases of retrobulbar inflammation. Two thirds of MS-associated unilateral ON will present with retrobulbar inflammation; the remaining will present with papillitis. Hemorrhages are rare in MS-associated papillitis.19 Like MS, NMOSD typically presents as retrobulbar inflammation. However, MOGAD commonly presents with papillitis in up to 86% of cases, and is more likely to exhibit severe disc edema and peripapillary hemorrhages. Whereas ON is typically unilateral in MS, bilateral ON is more common in MOGAD and NMOSD.19,21. Patients with inflammation in the chiasm or optic tract are also more likely to have a diagnosis of NMOSD.19
Therefore, patients with NMOSD lesions tend to have the most damage to the afferent visual pathway and tend to have the least post-inflammatory visual recovery .19,21-23 The recurrence of ON on an annualized basis is greater in MOGAD positive patients at a rate of 1.2 incidences per year compared to NMOSD and MS at 0.6 and 0.4 incidences per year respectively.20 Table 2 categorizes the likelihood of each demyelinating condition causing inflammation in the different areas of the visual pathway anterior to the lateral geniculate nucleus.19-21,24 Inflammation posterior to the lateral geniculate nucleus can occur, but typically with less significant afferent manifestations due to widely spaced distribution of axons when compared to tightly packed axons of the anterior visual pathway. Posterior inflammatory lesions are often incidental neuroimaging findings; these lesions may not always produce noticeable symptoms.24 Visual field defects may help identify possible locations of an inflammatory lesions involving the visual pathway. In terms of ON, however, only 33.6% of patients will present with a localized visual field defect, whereas the remaining 66% will present with diffuse visual field loss. Of the localized defects in ON, central/cecocentral defects are the most common, occurring approximately 29% of the time. Less frequently, ON presents as arcuate defects in 15.6%, altitudinal defects in 8%, and partial arcuate defects in 5%, all of which could mimic glaucoma.25 Chiasmal involvement typically presents as bitemporal hemianopsias, and optic tract lesions present as incongruous homonymous hemianopsias.19,23,25 Demyelinating lesions in the posterior visual pathway typically do not present with visual field defects, but rarely can demonstrate homonymous hemianopsias.24, 25
Additional ocualar testing
Clinical testing is critical to make an initial diagnosis of ON, monitor for interval change and serve as a biomarker for overall disease activity. Visual evoked potentials (VEP) can help differentiate between ON from demyelinating disease and other causes of optic neuropathy. In cases of ON from demyelinating diseases, the P100 latency is increased. In cases of optic neuropathy from ischemic, toxic or even compressive etiologies, the amplitude on a VEP is reduced while the P100 latency remains normal.19 OCT measurements in patients with demyelinating disease can provide evidence of acute and chronic neuronal degeneration. In acute papillitis, OCT of the peripapillary RNFL will demonstrate thickening due to active inflammation. If the lesion is more posterior, the OCT may initially appear normal. Whereas, in acute active demyelination, the disease burden can be imaged on MRI, chronic neuronal loss is more difficult to quantify as cerebral axonal atrophy is confounded by supportive cell loss. This pure axonal loss signifies disease progression, and in some cases, response to treatment. OCT is valuable because the retina and optic disc are the only CNS structures that can be seen in vivo non-invasively. Additionally, the ganglion cells and their axons, which make up the retinal nerve fiber layer are unmyelinated. Therefore, when the thickness of these layers is assessed with OCT, it is a more selective measure of axonal integrity. MS and NMOSD patients without a clinically identifiable episode of ON have an overall thinner RNFL and greater progressive RNFL loss than controls.26-28
In MS, the rate of progressive RNFL decline is similar whether or not the eye has had a known episode of ON.29 Since it is unclear if global neurodegeneration occurs in MOGAD, further studies are needed to determine whether there is progressive inner retinal thinning independent of ON events. OCT imaging can also identify macular changes, including microcystic edema associated with MS and MOGAD, as well as uveitis induced macular edema in MS, NMOSD, and MOGAD.30,31 MS and NMOSD patients can present with acute retinal inflammatory changes, including retinal vasculitis as well as post-treatment retinal necrosis.32-35 With the advent of OCT-angiography, studies have shown subclinical reduced choroidal and peripapillary vessel density in these patients.36-38 Another imaging modality eye care providers can consider is confocal microscopy of the cornea. Sensory nerves are prevalent in the cornea, and confocal microscopy has shown a reduction in their density in MS, paralleling the severity of the disease.39 Although we can see notable differences among demyelinating subtypes with in-office procedures, ophthalmic testing alone often cannot distinguish between these conditions. Additional work-up is critical to help with differentiation.
Diagnostic criteria
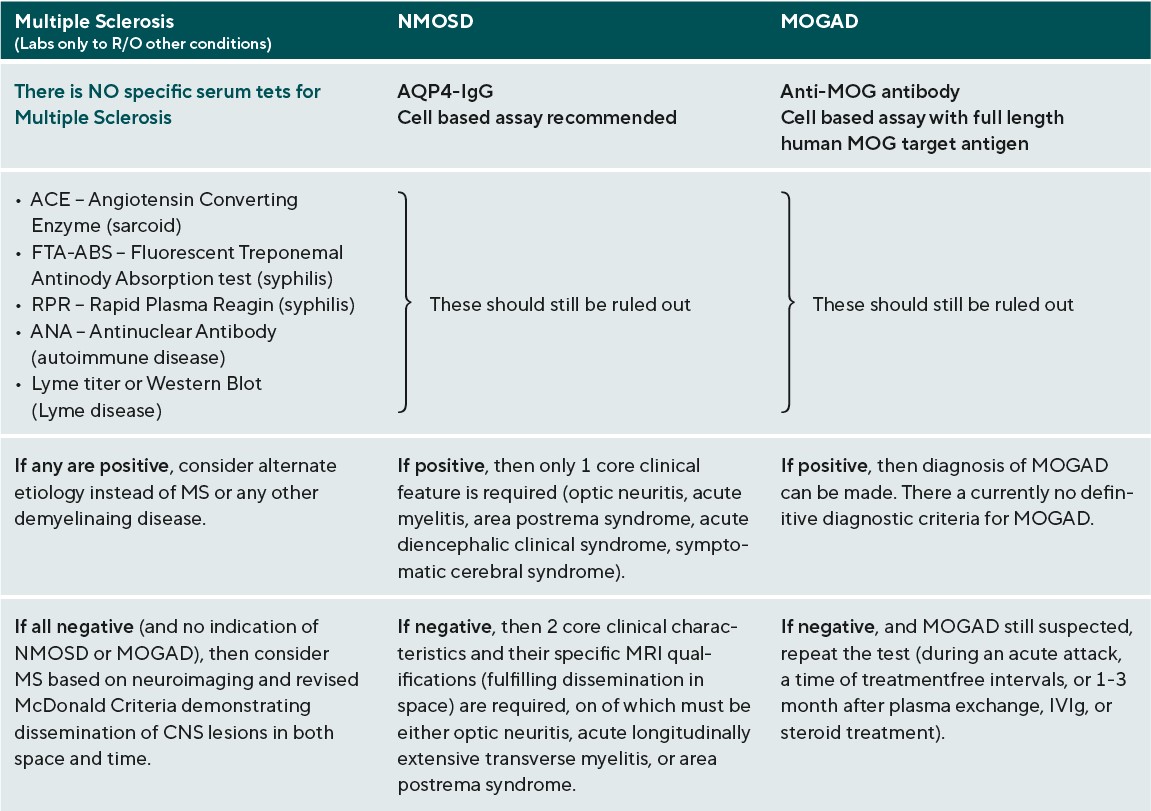
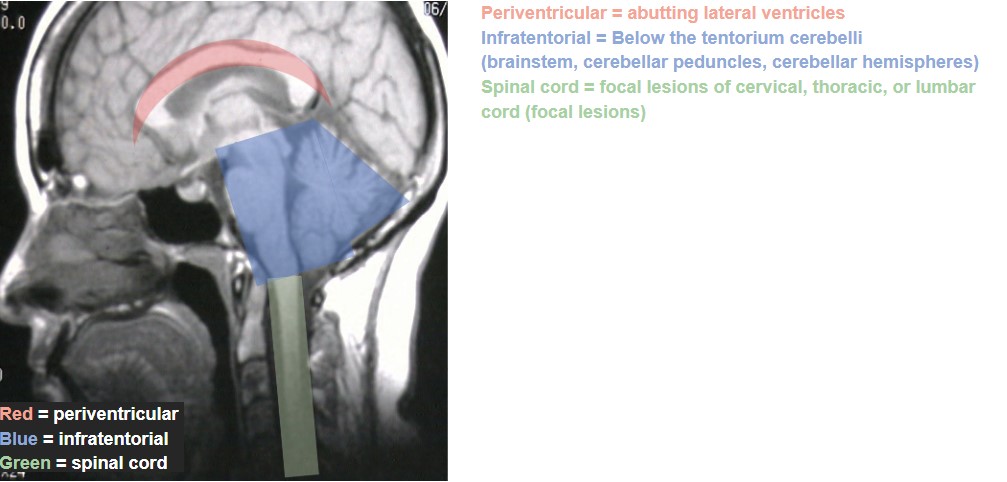
The work-up for any suspected demyelinating disease includes both serologic testing and neuroimaging of the brain, spinal cord, and possibly also of the orbits if the patient presents with acute ON. (Table 3)40-42 The specific findings of these tests are not themselves individually diagnostic, but do help to rule out other conditions and to differentiate among the demyelinating diseases. For MS, the role of serologic testing is mainly to rule out other conditions that could cause similar presenting features, such as syphilis, Lyme disease, sarcoid, and other autoimmune diseases. There is no specific blood test to confirm MS. Rather, the diagnostic criteria revolve around neuroimaging, and were revised most recently in 2017. The Revised McDonald Criteria require demonstration of dissemination of CNS lesions in both space and time. 40 In order to meet the diagnostic criteria for MS, a patient needs to experience 2 clinical attacks with either evidence of at least 2 lesions or evidence of 1 lesion with clear historical evidence of prior attack involving a lesion in a different location. Without this level of evidence, MS diagnosis cannot be confirmed until additional future evidence demonstrating dissemination in time and space can be made. Dissemination in time can be shown by an additional clinical attack, simultaneous presence of both enhancing and non-enhancing MS-typical MRI lesions, new T2 or enhancing MRI lesion compared to baseline scan, or CSF oligoclonal bands. Dissemination in space can be proven by an additional clinical attack implicating a different CNS site, or typical T2 lesions in at least 2 areas of the CNS (periventricular, cortical, juxtacortical, infratentorial, or spinal cord). (Figure 1)40,43 In a patient with steady progression of disease for 1 year since onset, CSF oligoclonal bands may be used, as may 1 or more MS-typical T2 lesions (periventricular, cortical, juxtacortical, or infratentorial), or at least 2 T2 spinal cord lesions to demonstrate dissemination in space. It is of note that clinically isolated ON, also known as clinically isolated syndrome, would not meet this criterion for MS diagnosis.40
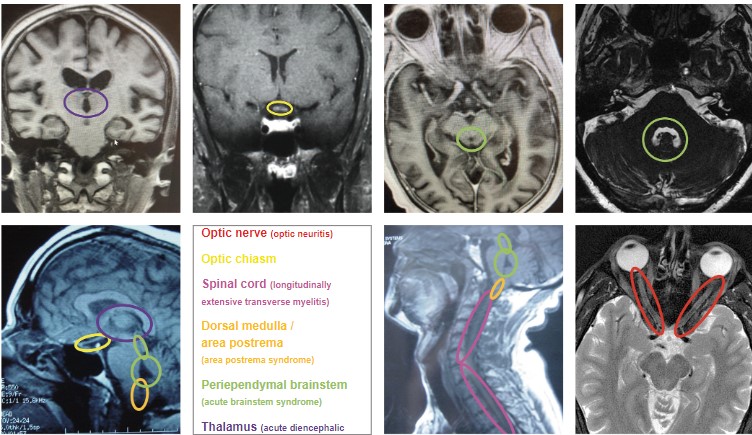
The adult diagnostic criteria for NMOSD were most recently revised in 2015 and include representative reference neuroimages. The serologic testing of greatest importance in NMOSD is AQP4-IgG, and a cell-based assay is strongly recommended. If serologic confirmation is proven, and other conditions are eliminated with lab testing, then only one core clinical feature is required to meet NMOSD diagnostic criteria. The core clinical features include: ON, acute myelitis, unexplained hiccups, or nausea / vomiting (area postrema syndrome), acute brainstem syndrome, symptomatic narcolepsy / acute diencephalic clinical syndrome (with typical MRI thalamic lesions), and symptomatic cerebral syndrome (with typical MRI brain lesions).(Figure 2)41 If there is no AQP4-IgG positivity, then 2 core clinical characteristics are required, one of which must be either ON, acute longitudinally extensive transverse myelitis, or area postrema syndrome. The 2 core clinical characteristics must fulfill dissemination in space. In addition, without AQP4-IgG confirmation, more specific MRI qualifications are needed. Acute ON must be accompanied by normal brain MRI or showing only nonspecific white matter lesions, or an orbital MRI must show a T2 hyperintense lesion or T1-weighted gadolinium enhancing lesion extending over >1/2 optic nerve length or involving the optic chiasm. The acute myelitis requires an associated intramedullary MRI lesion extending over 3 or more contiguous segments or 3 or more contiguous segments of focal spinal cord atrophy in patients with history of previous acute myelitis. Area postrema syndrome requires associated MRI dorsal medulla / area postrema lesions, and acute brainstem syndrome requires associated MRI periependymal brainstem lesions.41 There are currently no definitive diagnostic criteria for MOGAD. Diagnosis relies on the result of anti-MOG antibody testing. For that reason, it is critical that the test be performed appropriately for accurate analysis. Anti-MOG antibody testing should be performed on serum using the cell-based assay with full length human MOG as target antigen. Other methods including immunohistochemistry, ELISA, or Western blot testing are not recommended due to low specificity. If MOGAD is suspected, and the serum cell-based assay is negative, the test should be repeated during an acute attack, a time of treatment-free intervals, or 1-3 months after plasma exchange or treatment with IVIg or steroid infusions.42
Management / Prognosis
Management of demyelinating neurodegenerative conditions typically requires treatment of acute attacks followed by a chronic treatment regimen. ON is a characteristic manifestation of MS, NMOSD, and MOGAD. In the acute state, the recommended treatment for ON in demyelinating neurodegenerative condition is IV methylprednisolone (IVMP) 1000mg for 3-5 days followed by an oral taper. If the patient has a poor initial treatment response, the following treatments should be considered: repeat or increase IVMP to 2,000mg, add plasma exchange (PLEX) (in acute NMOSD and MOGAD) and /or add intravenous immunoglobulin (IVIG) (in acute NMOSD and MOGAD). MOGAD patients are likely to require a slower oral prednisone taper, over months, to avoid early relapses.44 As discussed earlier, both the characteristics and visual recovery of the associated ON vary among the demyelinating conditions. Generally, the visual recovery after a MS or MOGAD-related ON is favorable, but NMOSD-related ON tends to have a worse visual outcome.45 Long-term treatment of neurodegenerative demyelinating disease attempts to decrease the number of relapses by suppression or modulation of the immune system.
Multiple Sclerosis
Chronic MS treatments are disease-modifying by changing the body’s immune response from a pro-inflammatory to anti-inflammatory state. Older therapies target the T-cell inflammatory response (ex: glatiramer acetate) with later therapies attempting to inhibit lymphocyte access to the CNS (ex: natalizumab) and the most recent therapies work to decrease the B-cell response (ex: ocrelizumab).46 Whereas initial therapies were injectable, more recent treatment options include orals and infusions. Historically, over time, RRMS showed generalized progression of disability leading to progressive MS over a period of approximately 19 years.46 With treatment, episodes of relapse have significantly declined, but over time still show an underlying silent progression. This has led to the recommendation of aggressive treatment early in the disease course before permanent disability is evident.46
NMOSD and MOGAD
The disease courses of NMOSD and MOGAD are antibody-driven; thus, their preventative treatments are primarily immunotherapy. Relapses can occur in both NMOSD and MOGAD, with the highest rate of relapse in seropositive NMOSD.45,47 Commonly used drugs for chronic treatment include: rituximab (decreases CD20 B cells), mycophenolate mofetil (inhibits B and T cells) and azathioprine (inhibits proliferation of lymphocytes). Other biologics used in some cases include: tocilizumab, eculizumab, satralizumab, inebilizumab and IVIG.47 Long-term large clinical trials need to be concluded to determine a standard of care, but analysis thus far shows that rituximab is most effective in NMOSD treatment.48 Long-term treatment of MOGAD is still being studied, but in many cases, the condition has been monophasic and long-term treatment may not be necessary, particularly in pediatric populations.49
Potential visual concerns with treatment
Fingolimod is the first sphingosine 1-phosphate receptor modulator treatment used for MS, and has been associated with macular edema, typically within four months of starting the treatment. Macular edema occurs in less than 5% of MS patients but there is increased risk in patients with a history of uveitis or diabetes. It is recommended to screen patients prior to or within one week of starting treatment and again in four months.50 All subsequent MS medications of the same class have a similar risk of macular edema; these include siponimod, ponesimod, and ozanimod. Alemtuzumab is an anti-CD52 monoclonal antibody infusion treatment for MS. Alemtuzumab-associated thyroid eye disease occurs in less than 2% of MS patients but can occur at any time during their infusion treatments.50 A concern about treating patients with immunomodulating therapy is the possibility of developing progressive multifocal leukoencephalopathy (PML) in patients who are JC virus positive.
PML is incurable, can cause severe disability and is potentially fatal. Thus, patients are screened for the JC virus prior to starting many of the therapies used to treat demyelinating neurodegenerative conditions. Some of the medications which have been associated with PML and require pre-screening include: mycophenolate mofetil, natalizumab, and rituximab for NMOSD and MOGAD, as well as, dimethyl fumarate, fingolimod and ocrelizumab, among many others for MS. PML has been linked with medications from varying classes and with all routes of administration. Many types of visual dysfunction may result from PML including: homonymous hemianopias, higher order processing dysfunction, nystagmus and diplopia.50 None of the older injectable MS treatments (interferon β1a, interferon β1-b, glatiramer acetate) have been linked with PML.
Conclusion
Eye care providers are a vital part of the care team and are often the first to suspect demyelinating disease based on the presenting ocular features, most commonly ON. Understanding similarities and differences among these demyelinating diseases is vital to ensure prompt appropriate initial work-up, referral, diagnosis, and treatment, which can vary among these conditions. (Table 4)1-14,19-24,44-50 Eye care providers remain essential in the ongoing management of patients with demyelinating diseases as well, working closely with neurology not only during acute flare-ups, but also for assessment of stability. Use of serial OCTs can help monitor for interval progressive neurodegeneration, which can in turn assess effectiveness of current treatment plans. Eye care providers need to remain updated with the latest scientific discoveries, diagnostic criteria, and management options regarding these neurodegenerative demyelinating diseases in order to provide the best possible care to their patients.
Conflict of Interest
None of the authors has any conflict of interest to disclose related to this manuscript.
