Therapieansätze bei Netzhauterkrankungen

Frau Professor Wohl, Sie lehren und forschen an der State University of New York, College of Optometry an dem nach Ihnen benannten Wohl Lab des Department of Biological & Vision Science. Wir wissen heute, dass es einen Zusammenhang zwischen Gesundheit und Funktion des visuellen Systems und einer korrekten Genexpression gibt. In diesem Kontext untersuchen Sie unter anderem die Rolle von microRNAs im Zusammenhang mit der Reaktion von Gliazellen bei Erkrankungen der Netzhaut. Würden Sie bitte unseren Lesern einmal Ihre aktuellen Forschungsaktivitäten beschreiben, und warum Fehlfunktionen der Gliazellen Ursache für neurodegenerative Erkrankungen der Netzhaut verantwortlich sein können?
Neben den Nervenzellen sind die Gliazellen der zweite wesentliche Zelltyp in allen Teilen des Zentralnervensystems, einschließlich der neuralen Netzhaut. Die Gliazellen haben fundamentale Aufgaben und Funktionen zu denen die Versorgung der Nervenzellen mit Nährstoffen, Recycling von Neurotransmittern, Sicherstellung der Homöostase (Wasser und Ionenausgleich) sowie Stabilität und Schutz des Gewebes gehören. Von daher geht ein Verlust der Gliafunktion mit fatalen Konsequenzen für die Netzhaut einher. Es gibt in der Netzhaut zwei wesentliche Arten von sogenannten Makrogliazellen: die Astrozyten und die Müllergliazellen. Die Astrozyten machen nur etwa 0,5 Prozent der gesamten Netzhaut-Zellpopulation aus und sind in der Nervenfaserschicht der Netzhaut zu finden.
Eine wesentliche Aufgabe der retinalen Astrozyten ist die Bildung der Blut-Retina- Schranke, die verhindert, dass Erreger oder größere Moleküle in das Gewebe eindringen können (Schutzfunktion). Astrozyten wandern während der Entwicklung vom Gehirn über den Nervus opticus in die Netzhaut ein und entstammen einer nicht-retinalen Zellquelle. Astrozyten spielen vor allem bei einem Glaukom eine wesentliche Rolle. Die Müllergliazellen hingegen sind die dominierenden Makroglia der Netzhaut (etwa fünf Prozent der Netzhaut- Zellpopulation) und nur in der Netzhaut zu finden. Sie wurden von dem deutschen Anatom Prof. Heinrich Müller erstmals beschrieben und sind nach ihm benannt. Ein sehr wichtiger Fakt ist, dass Müllergliazellen aus einer retinalen Vorläuferzelle (einer Art Stammzelle) hervorgehen, die während der Retinogenese alle sechs retinalen Nervenzellen bildet. Die Bildung der Netzhautzellen erfolgt dabei einer hoch-konservierten Sequenz, die in allen Wirbeltieren zu finden ist: in der ersten Phase (vor der Geburt) entstehen die Ganglienzellen, Horizontalzellen, Zäpfchen und Amakrinzellen. In der zweiten Phase (nach der Geburt) die Bipolarzellen, Stäbchen und zum Schluss die Müllerglia. Um es etwas bildhafter auszudrücken, die Müllergliazellen sind die kleinen Geschwister der Netzhautzellen und das macht diese Zellen sehr besonders und für regenerative Strategien sehr interessant.
Wir wissen heute, dass Gliazellen bei Netzhauterkrankungen eine wesentliche Rolle spielen. Es gibt sogar Hinweise darauf, dass Fehlfunktionen der Müllerglia zu einer Form von Retinitis pigmentosa führen. Wie das genau geschieht, ist aber noch nicht erforscht. Fehlfunktionen von Gliazellen können durch genetische Veränderung in den Gliazellen selbst hervorgerufen werden (meist eine Mutation der Gliagene) oder aber durch die Veränderung der Zellen aufgrund von einer veränderten Umgebung. Letzteres ist bei jeder Art von Verletzung der Fall und kann unterschiedliche Ausmaβe annehmen. Gliazellen, die sich bei Verletzung und/oder Erkrankung des Gewebes verändern, sind nicht mehr in der Lage, ihrer eigentlichen Funktion nachzugehen und das verschlimmert den Zustand des Gewebes. Herr Professor Dr. Andreas Reichenbach der Universität Leipzig hat die Rolle der Müllergliazellen in der gesunden und erkrankten Netzhaut über Jahrzehnte studiert und seine Erkenntnisse in einem wundervollen Buch zusammen mit Professor Dr. Andreas Bringmann publiziert, das ich jeden nur empfehlen kann, der Interesse für Müllerglia und Netzhauterkrankungen hat.
Es ist bekannt, dass Gliazellen bei Verletzung des Gewebes zunächst neuroprotektiv sind sowie das geschädigte Gewebe vom gesunden Gewebe isolieren. Die Gliazellen unterziehen sich dabei molekularer Veränderungen, die wiederum Veränderungen in der Morphologie und Funktion der Zellen mit sich bringen. Dieser Prozess wird Gliose genannt. Der Verlust der physiologischen Funktion (Nervenzellversorgung, Neurotransmitter-Recycling, Wasser-Ionen-Ausgleich) hat jedoch verheerende Auswirkungen auf das noch gesunde Gewebe. Hinzu kommt, dass bei sehr schweren und andauernden Schädigungen die Gliazellen beginnen, neurotoxische Moleküle zu bilden und freizugeben. Diese toxischen Moleküle führen dann zum Tod von Nervenzellen, die nicht direkt geschädigt wurden. Das wird oft als sekundärer Nervenzelltod bezeichnet. Zudem können Müllergliazellen immunkompetente Zellen (Mikrogliazellen, Leukozyten, Makrophagen etc.) aktivieren und rekrutieren, die zu einer Entzündungsreaktion führen. Dieser Prozess der Gliose ist sehr komplex und viele Fragen hinsichtlich möglicher verschiedener Gliosearten/ -stadien und potenzieller Manipulationsstrategien zur Abschwächung oder sogar Verhinderung von Gliose sind nach wie vor unbeantwortet. Fakt jedoch ist, die Gliazellen spielen eine maßgebliche Rolle bei Netzhauterkrankungen.
Ich hatte anfangs erwähnt, dass es nicht auszuschließen ist, dass Gliafehlfunktionen sogar die Ursache für Netzhauterkrankungen sein können, ohne dass eine neuronale Schädigung vorliegt. Es gibt in der Tat Hinweise darauf, dass eine Gliafehlfunktion zu einer Form von Retinitis pigmentosa führen kann und ich selbst hatte 2017 ein ähnliches Resultat erhalten, als wir ein sehr künstliches Experiment durchgeführt hatten. Ich war damals Postdoc im Labor von Professor Thomas Reh (University of Washington) in Seattle. Unsere Fragestellung war, ob microRNAs eine Rolle in der Funktion von Müllergliazellen spielen. MicroRNAs waren damals eine relativ neu-identifizierte Molekülgruppe mit unbekannter Funktion für retinale Zellen. MicroRNAs sind regulatorische Moleküle und bei der Entstehung von Krebs (Tumorbildung) involviert. Sie werden vor allem in der Krebsforschung bereits als Indikatoren/Biomarker genutzt und könnten auch als therapeutisches Mittel von großer Bedeutung sein. Gründe hierfür sind zum einen ihre kleine Größe. Kleine Moleküle können relativ barrierefrei durch den Körper reisen und können somit zum Beispiel in Körperflüssigkeiten wie Blut nachgewiesen werden (Indikatorfunktion). Kleine Moleküle können auch sehr gut in Zellen eingebracht werden und somit mögliche Ungleichgewichte auszugleichen (Therapieansätze). Ein weiterer Vorteil ist, dass Veränderungen der microRNA-Konzentration (An- oder Abwesenheit) bereits messbar ist, bevor es zu zellulären oder gar histologischen Veränderungen kommt. Dieses Ergebnis haben wir in der Netzhaut in unserem Versuch gesehen und es wurde auch von anderen Forschern anderer Fachgebiete bestätigt. Das heißt, theoretisch könnte eine Erkrankung erkannt werden, bevor es zum Zellverlust kommt, sofern sich diese Erkrankung erst später im Leben eines Patienten etabliert. Bei sehr vielen Netzhauterkrankungen ist dies jedoch der Fall.
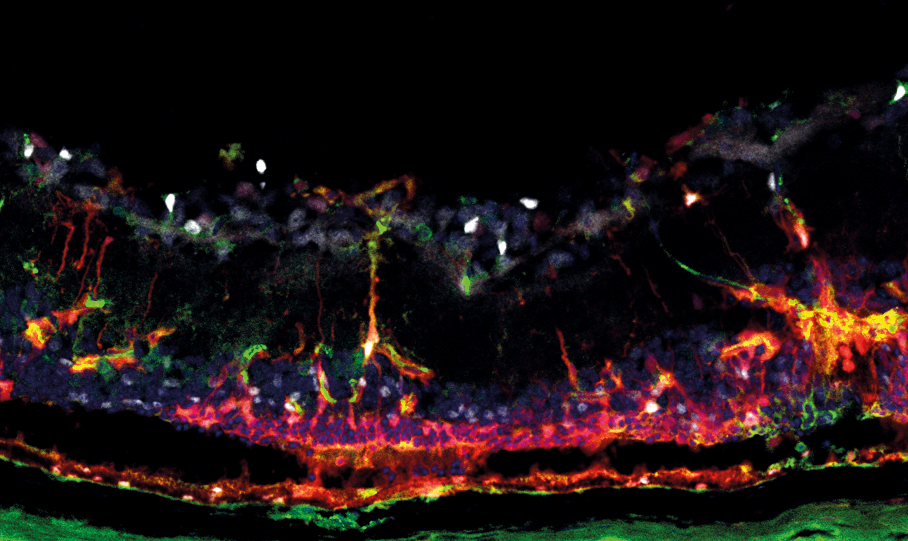
Um nun die Frage zu beantworten, ob microRNAs für die Funktion von Müllergliazellen relevant sind, haben wir uns eines Mausmodells bedient, dass uns erlaubte, microRNAs in den Müllergliazellen zu eliminieren/reduzieren. Wir arbeiten mit genetisch veränderten Mäusen als Modellsystem, die Versuche dieser Art ermöglichen. Da fast alle microRNA-Sequenzen in Maus und Menschen identisch (hoch-konserviert) sind, ist die Wahrscheinlichkeit groβ, dass unsere Forschungsergebnisse von der Maus erfolgreich auf den Menschen übertragen werden können (Stichwort Translational Research).
Wir hatten ein Jahr zuvor herausgefunden, dass Müllergliazellen ein definiertes Set von microRNAs haben, das sich von dem der Nervenzellen und Astrozyten unterscheidet. Wir fanden ebenfalls heraus, dass die Menge/Anzahl dieser Müllerglia-microRNAs mit zunehmendem Alter ansteigt. Dies konnten wir durch eine neue Methode analysieren, die zu diesem Zeitpunkt entwickelt wurde und uns erlaubte, die Anzahl der microRNA-Moleküle zu bestimmen. Wir vermuteten, dass diese Müllerglia-microRNAs eine spezifische Funktion haben müssen. Wir eliminierten die microRNAs in den Müllergliazellen der Tiere (keine Nervenzellen waren beeinträchtigt) und machten sehr interessante Beobachtungen. Sechs Monate nach der Manipulation der Gliazellen fanden wir einen Phänotyp, der an Retinitis pigmentosa erinnert. Es war ein drastischer Verlust von Photorezeptoren zu finden, obwohl das Gewebe (Photorezeptoren) nicht geschädigt wurde. Das konnte nur bedeuten, dass die Nervenzellen aufgrund der veränderten Gliazellen absterben. Darüber hinaus konnten wir keine Müllerglia-Gliose finden. Das ließ vermuten, dass Müllerglia-microRNAs für Glioseprozesse wesentlich sind.
Diese Arbeit bildet nun ein wesentliches Fundament für meine eigene Forschung an der State University in New York (SUNY) mit der Fragestellung: Wie können wir die physiologische Funktion der Müllergliazellen bei Verletzung oder Erkrankung aufrechterhalten, so dass es nicht zu einer zunehmenden Verschlechterung des Gewebes kommt (zum Beispiel zusätzlicher Nervenzellverlust oder Entzündungsreaktionen). Eine Studie, die in Zusammenarbeit mit meinem früheren Mentor Dr. Thomas Reh 2020 erschien, zeigt, dass microRNAs in den Gliazellen nach Schädigung der Netzhaut ebenfalls reduziert sind. Das war ein unerwarteter, aber sehr interessanter Befund. Wir versuchen nun den gemeinsamen Nenner zu finden, indem wir unser künstliches Modell der microRNA-Eliminierung mit dem Lichtschaden-Modell sowie einem Retinitis pigmentosa Mausmodell vergleichen. Unsere Hypothese ist, dass wir die verloren gegangenen microRNAs künstlich dem Gewebe (den Müllergliazellen) zuführen, die physiologische Funktion der Zellen wiederherstellen können und somit den Krankheitsverlauf abschwächen können.
In Ihrer gemeinsamen Publikation „Stimulation of functional neuronal regeneration from Müller glia in adult mice“, die Sie zusammen mit Nikolas L. Jorstad et al. in der Zeitschrift Nature 2017 publiziert haben, beschäftigen Sie sich gerade mit dem Verlust von Netzhautneuronen und einer neuronalen Regeneration von Müllerzellen bei Mäusen. Hier handelt sich sicherlich um eine Grundlagenforschung. Welche Erkenntnisse konnten Sie in dieser Studie gerade im Hinblick von möglichen Gentherapien bei Erkrankungen der Netzhaut gewinnen?
Das Nature-Paper vom Reh Labor hatte 2017 zum ersten Mal zeigen können, dass nach experimenteller Läsion, Müllergliazellen der adulten Maus zu funktionalen neuen Nervenzellen umprogrammiert werden können. Das war eine bahnbrechende Entdeckung, da in der Maus und allen Säugetieren einschließlich Mensch, keine natürlichen regenerativen Ereignisse im Zentralnervensystem existieren. Hinzu kommt, das bis dato die Frage offen war, ob neue Nervenzellen in einem geschädigten Gewebe überhaupt überleben und sich funktionell integrieren können. Die Antwort ist JA und das ist sehr vielversprechend.
In dieser Arbeit wurde sich eines Mausmodells bedient, das einen Transkriptionsfaktor überexprimiert, genannt ASCL1, der die Gliazellen zu unreifen stammzellartigen Zellen umprogrammiert. ASCL1 ist ein Gen, das während der Netzhautentwicklung aller Wirbeltiere einschließlich des Menschen entscheidend ist und bei der Netzhautregeneration im ausgewachsenen Fisch eine fundamentale Rolle spielt. Fische können im Gegensatz zu Säugern ihre Netzhaut vollständig regenerieren. Das erfolgt mittels ihrer Müllergliazellen, die als natürliche regenerative Zellquelle dienen.
Die Idee ist nun, humane Müllerglia zur Regeneration zu befähigen. Dazu müsste mittels Gentransfer (sehr wahrscheinlich über virale Vektoren), das ASCL1 Gen in humane Müllergliazellen eingebracht werden. Es gibt noch sehr viel Arbeit in diesem Forschungsbereich, aber erste Schritte sind gemacht. Diese Zellersatztherapie könnte dann ubiquitär Einsatz finden, unabhängig von der Art der Netzhauterkrankung.
In diesem Kontext habe ich auf PubMed eine ganz aktuelle Publikation von Ihnen mit dem Titel „View on microRNAs as a potential tool to fight blindness: focus on Müller glia and gliosis“ in der Zeitschrift Neural Regeneration Research aus dem Jahr 2022 gefunden. Gerade den Abschnitt „microRNAs als potenzielles therapeutisches Mittel zur Abschwächung der Gliose“ fand ich sehr interessant. Können Sie unseren Lesern hierzu vielleicht noch Einiges erläutern.
Wie eingangs erwähnt, geht jede Netzhauterkrankung mit Gliose einher. Der Verlauf und die Schwere der Gliose können sehr unterschiedlich sein, aber die Gliazellen durchlaufen immer ähnliche Veränderungen. Der Artikel von 2022 ist ein Viewpoint (Sichtweise) und bezieht sich vor allem auf die Erkenntnisse von den Müllerglia-microRNA Studien von 2017 und 2020 sowie andere Arbeiten, die zeigen, dass microRNAs Schlüsselmoleküle der Gliose regulieren und folglich ein potenzieller Ansatzpunkt für therapeutische Intervention bieten.
MicroRNAs sind eine relativ neue Molekülgruppe. Sie wurden 1993 erstmals beschrieben und heißen seit 2001 microRNAs. Sie gehören zu den sogenannten nicht-kodierenden RNAs, das ist eine Familie von regulatorischen RNAs, die nicht zu einem Protein umgesetzt werden. Und diese RNA Familie wird aufgrund verbesserter Methodik zunehmend größer, das heißt immer mehr Moleküle werden entdeckt und identifiziert.
MicroRNAs interagieren auf dem Level der messenger RNA (oder kurz mRNA) und Verhindern den Umsatz dieses Transkripts zum Protein, indem sie an die mRNAs binden (diese somit blockieren). Daher werden microRNAs oft in das Gebiet der Epigenetik eingestuft. Dieses Blockieren mag zunächst negativ klingen, ist aber ein Standardmechanismus, der in allen Zellen stattfindet und wesentlich für Zellidentität und -funktion ist. MicroRNAs sind, wie der Name annehmen lässt, und wie bereits erwähnt, sehr klein (18-24 Basenpaare) und können somit sehr leicht in Zellen eingebracht werden. Sie haben zudem mehrere Zielmoleküle (Ziel-mRNAs), was einen verstärkten Effekt mit sich bringen kann. Sie können kombiniert und dosiert werden und sowohl temporäre als auch kontinuierliche Behandlung ist theoretisch möglich.
Wir wissen, dass Müllergliazellen ein spezifisches Set an microRNAs haben, und dass der Verlust von microRNAs in Müllergliazellen zur Beeinträchtigung der Funktion der Gliazellen führt. Die Folge sind neurodegenerative Ereignisse, die mit Verlust der Sehkraft einhergehen. Die erwähnte Studie aus meinem Labor in Zusammenarbeit mit meinem früheren Mentor Tom Reh, (publiziert 2020), zeigte, dass reaktive (gliotische) Müllergliazellen die meisten ihrer microRNAs verlieren (80 Prozent Verlust). Wir beobachten dieses unerwartete Phänomen, das teilweise unserem künstlichen microRNA Eliminierungs-Experiment gleicht, nach Schädigung der Netzhaut mit einem Lichtschaden-Modell. Das Lichtschaden-Modell ist ein Modell, das zu massiven Photorezeptorzellverlust führt und einen Phänotyp aufzeigt, der degenerativen Erkrankungen, wie zum Beispiel die der altersbedingte Makuladegeneration (AMD), ähnelt. Da microRNAs die Umsetzung von mRNA zu Protein hindern, haben wir uns dann die Transkripte (mRNA) angesehen, die in reaktiven Gliazellen erhöht waren, also die potenziellen Zielmoleküle der microRNAs. Die Hypothese ist, dass die Transkripte (oder deren translatierte Proteine), die in normalen Müllergliazellen nicht vorhanden sind, aber nach Läsion erscheinen, hochreguliert sind, die Moleküle sind, die normalerweise von den Müllerglia-microRNAs blockiert werden. Da die microRNAs in reaktiven Müllerglia verloren gehen und nicht mehr blockieren können, sehen wir daher eine Hochregulation dieser Zielmoleküle. Es liegt nun nahe, herauszufinden, ob das Wiedereinbringen dieser verloren gegangenen microRNAs in die Müllergliazellen den physiologischen Zustand der Zellen wieder herbeiführen kann. Somit wäre es zumindest theoretisch möglich, Gliose zu regulieren/abzumildern. Wie anfänglich ausgeführt, könnte eine verminderte Gliose zu weniger Zellverlust führen (ausbleibender sekundärer Zelltod). Folglich wäre das Gewebe gesünder und sehr wahrscheinlich auch weniger funktionell beeinträchtigt, was im Prinzip ein Hinauszögern, vielleicht sogar in Aufhalten der Verschlechterung der Sehkraft bedeuten würde. Wir stehen aber noch am Anfang dieser Forschung und viele Fragen und Aspekte sind noch unklar, beziehungsweise hängt sehr viel davon ab, was experimentell umsetzbar ist, gerade was den Molekültransfer angeht.
Wir wissen heute, dass genetische Faktoren sowohl bei monogenetischen Netzhauterkrankungen, wie zum Beispiel der bekannten Retinitis pigmentosa oder anderen hereditären Netzhautdystrophien, aber auch bei multigenetische Erkrankungen der Netzhaut mit genetischen Risikofaktoren, wie zum Beispiel einer altersbedingten Makuladegeneration (AMD), eine große Rolle spielen. Wie weit ist der Stand der diesbezüglichen Forschung heute gerade im Bereich der in der Regel monogenetisch verursachten Netzhautdystrophien?
Durch bahnbrechende Methoden wie zum Beispiel Genomsequenzierung oder Einzelzell- RNA Sequenzierung (engl. single cell RNA-seq) konnten und können sehr viele genetische Ursachen für Netzhauterkrankungen identifiziert werden. Ich bin keine Klinikerin, aber ich kann mir vorstellen, dass die Liste der Gen-Kandidaten stetig zunimmt. Wenn die Erkrankung auf einer angeborenen Mutation basiert, so wird durch Gentherapie versucht, den Schaden auszugleichen. Dazu wird das defekte Gen (fehlendes Genprodukt) durch Einbringen eines funktionalen Gens ersetzt. Diese Gene sind dann quasi Zusatz-DNA in den Zellen, die DNA des Patienten bleibt dabei unbeeinträchtigt. Soviel ich weiß, ist die Erfolgsrate jedoch nicht sehr hoch. Das Einbringen von Zusatz-DNA ist sehr kompliziert, beruht meist auf viralen Vektoren und kann Entzündungsreaktionen hervorrufen.
Durch die Methode des CRISPR-Cas9 (Nobelpreis für Chemie 2020) haben sich seit 2018 nun aber ganz neue Möglichkeiten aufgetan, da durch diese „Gen-Schere“ theoretisch kranke Gene in der DNA des Patienten herausgeschnitten werden könnten und durch funktionale neue Gene ersetzt werden könnten. Das ist zumindest die Theorie und Hoffnung. Die Methode ist allerdings noch relativ neu und es gibt noch sehr viele riskante Nebeneffekte, die es erst zu eliminieren gilt, da dieses Werkzeug die DNA des Patienten verändert und es fatal wäre, wenn die falschen Regionen editiert werden würden. Forscher weltweit arbeiten jedoch daran, dieses Werkzeug zu perfektionieren und die Fortschritte sind beachtlich und bemerkenswert.
Aufgrund der enormen phänotypischen Vielfalt der Netzhautdystrophien ist die Diagnosestellung dieser Erkrankungen nicht immer einfach. Inwieweit hilft hier eine Gensequenzierung gerade bei der Diagnostik solcher Erkrankungen?
Es gibt sehr unterschiedliche Methoden, die helfen können, die Ursachen für bestimmte Phänotypen und Krankheitsbilder zu finden. Diese Methoden entwickeln sich stetig weiter und erlauben mittlerweile sehr detaillierte Analysen. Allerdings sind diese Methoden sehr kostenintensiv. Es gibt die Möglichkeit der DNA-Sequenzierung die mittlerweile erlaubt, das gesamte Genom eines Organismus zu sequenzieren, um zum Beispiel eine Mutation zu finden. Diese DNASequenzierung zeigt Veränderungen in der DNA Sequenz auf und das kann hilfreich sein, um einen Diagnoseverdacht zu untermauern. Dann rückt dieses spezielle Gen in den Fokus der Forschung.
Es gibt aber auch sehr viele Erkrankungen, die nicht unbedingt einen genetischen Hintergrund haben, sondern bei denen die Epigenetik eine Rolle spielt. Epigenetik ist ein relativ neues Gebiet, das immer mehr an Bedeutung gewinnt. Hier spielen Umweltfaktoren (UV-Licht), Ernährung, Gewohnheiten (Rauchen) etc. eine Rolle. Diese Faktoren können Einfluss auf die Struktur der DNA haben oder die Umsetzung des Genprodukts regulieren (Stichwort microRNAs). Es gibt aber, zumindest nach meinem Kenntnisstand, momentan noch keine Möglichkeit, epigenetische Analysen für Diagnostik im Patienten heranzuziehen, da die epigenetische Landschaft meist gewebespezifisch ist und folglich Gewebeproben (Netzhautgewebe) erforderlich wären. Da Gewebeproben der Netzhaut nicht möglich sind, muss auf Modellsysteme zurückgegriffen werden, um Erkenntnisse dann auf den Patienten übertragen zu können. Aus diesem Grund ist Grundlagenforschung so essenziell.
Vielleicht kurz ein paar Worte über den Einfluss von Epigenetik auf Erkrankungen. Gene können nur gelesen (transkribiert/ umgesetzt) werden, wenn die DNA „entpackt“ ist. Falls DNA als Knäuel vorliegt, ist sie nicht lesbar. Die Veränderung der DNA-Struktur in einen lesbaren oder unlesbaren Zustand kann spontan geschehen und bedeutet, die Gene können völlig intakt sein, aber nicht gelesen werden. Das Ergebnis ist ähnlich einer echten Mutation (Gendefekt), das Genprodukt wird nicht gebildet, aber die Ursache ist eine andere. Das kann auch anders herum geschehen, Gene, die normalerweise inaktiv sind (Knäuel) beziehungsweise sein müssen, werden entpackt und gelesen. In diesem Fall kann ein zusätzliches Genprodukt existieren, das der Zelle schaden und zur Erkrankung des Gewebes führen kann. MicroRNAs werden oft auch in die Epigenetik-Kategorie eingestuft. Falls eine microRNA in einer Zelle aktiv wird, in der sie normalerweise nicht aktiv ist, wird diese „falsch“ existierende microRNA die Bildung des Proteins ihrer Zielmoleküle unterbinden. Das Ergebnis ist ebenfalls ein fehlendes Genprodukt. Diese Ursachen des fehlenden Genprodukts können dann nicht mittels DNA-Sequenzierung ermittelt werden. Die derzeit verfügbaren Methoden, die eine Analyse auf Transkriptionsniveau erlaubt, ist die RNA-Sequenzierung. Eine RNA-Sequenzierung kann auf microRNA Niveau erfolgen und somit „neue“ oder fehlende microRNAs im Gewebe aufzeigen. Eine Methode, die eine DNA Strukturanalyse ermöglicht, ist die sogenannte ATAC-Sequenzierung (die Abkürzung steht für Assay for Transponase-Accessible Chromatin), die aufzeigt, ob DNA lesbar ist (zugängliches Chromatin). Beide Methoden sind mittlerweile sogar auf Einzelzellniveau durchführbar, aber sehr kostenintensiv und erfordern relativ große Gewebeproben. Jedoch haben diese Einzelzell-Methoden gezeigt, dass selbst Zellen einer Zellpopulation sehr heterogen sein können. Diese Methoden sind für die Diagnostik eines Patienten derzeit eine weniger relevante Technik, dennoch werden sie im Tiermodell, Organoid-Systemen und postmortem Geweben eingesetzt, um Krankheitsverläufe zu charakterisieren und diese Erkenntnisse dann auf Patienten übertragen zu können.
Frau Professor Wohl, wir haben nun viel über die Grundlagenforschung im Kontext genetischer Aspekte von Netzhauterkrankungen erfahren. Wie sehen Sie die Möglichkeiten aktueller und zukünftiger Gentherapien bei erblichen Netzhauterkrankungen? Für welche dieser Erkrankungen gibt es schon gentherapeutische Ansätze – für welche Erkrankungen erwarten Sie in der Zukunft Therapieoptionen?
Hier kommt es sicherlich darauf an, wann die Erkrankung auftritt und es bedarf sehr ausgereifter Methodik. Grundlagenforscher arbeiten daher daran, letzteres maßgeblich voranzutreiben.
Erkrankungen, die bei erwachsenen Patienten auftreten, sind meist schwieriger zu behandeln. Erwachsenes Gewebe ist weniger plastisch und ein Molekültransfer ist sehr schwierig. Hinzu kommt, dass oftmals Erkrankungen erst sichtbar werden, wenn bereits ein größerer Schaden (signifikanter Zellverlust) vorliegt. Daher sind die Erfolgsquoten für Gentherapie relativ gering, wenngleich nicht Null. Auch die Genschere ist sehr erfolgversprechend, wirft momentan aber noch sehr viele ethische Fragen auf. Eine der größten Hürden ist das erfolgreiche Einbringen von Molekülen in die korrekten (geschädigten) Zellen. Und selbst die Genschere beruht auf Molekültransfer, da die Maschinerie ebenfalls in die Zielzellen eingebracht werden muss. Ein weiteres Problem ist, die Fracht (Austausch-Gen oder Genschere) in die korrekten (defekten) Zellen zu bekommen. Eine neue Richtung für Therapieansätze sind die bereits erwähnten extrazellulären Vesikel oder Nanopartikel. Extrazelluläre Vesikel sind ein natürliches Transportsystem, das in unseren Zellen zu finden ist. Nanopartikel sind künstlich erzeugte Transportmoleküle, die, wie auch die Vesikel, zellspezifisch agieren könnten. Somit wären zelltyp-spezifische Therapien möglich. Aber selbst wenn noch bestehende Zellen „gerettet“ werden können, hilft Gentherapie oder die Genschere nicht, um bereits verloren gegangene Zellen zu ersetzen. Daher ist die Zellersatztherapie wesentlich. Kurzum, ich gehe davon aus, dass eine Kombination von verschiedenen Therapieansätzen sehr wahrscheinlich am erfolgreichsten sein wird – für alle Formen von Netzhauterkrankungen.